Department of Molecular Biology and Biochemistry, University of California Irvine, Irvine, CA, 92697, USA.
Department of Developmental and Cell Biology, University of California Irvine, Irvine, CA, 92697, USA.
Breast Cancer Res. 2021 Sep 27;23(1):93. doi: 10.1186/s13058-021-01468-x.
Cancer metastasis is a complex process involving the spread of malignant cells from a primary tumor to distal organs. Understanding this cascade at a mechanistic level could provide critical new insights into the disease and potentially reveal new avenues for treatment. Transcriptome profiling of spontaneous cancer models is an attractive method to examine the dynamic changes accompanying tumor cell spread. However, such studies are complicated by the underlying heterogeneity of the cell types involved. The purpose of this study was to examine the transcriptomes of metastatic breast cancer cells using the well-established MMTV-PyMT mouse model.
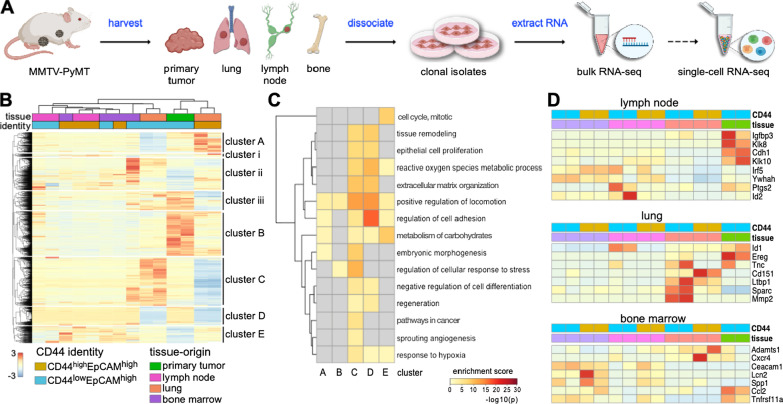
Organ-derived metastatic cell lines were harvested from 10 female MMTV-PyMT mice. Cancer cells were isolated and sorted based on the expression of CD44/EpCAM or CD44/EpCAM surface markers. RNA from each cell line was extracted and sequenced using the NextSeq 500 Illumina platform. Tissue-specific genes were compared across the different metastatic and primary tumor samples. Reads were mapped to the mouse genome using STAR, and gene expression was quantified using RSEM. Single-cell RNA-seq (scRNA-seq) was performed on select samples using the ddSeq platform by BioRad and analyzed using Seurat v3.2.3. Monocle2 was used to infer pseudo-time progression.
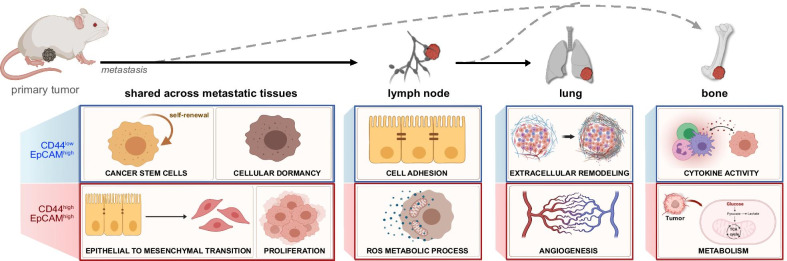
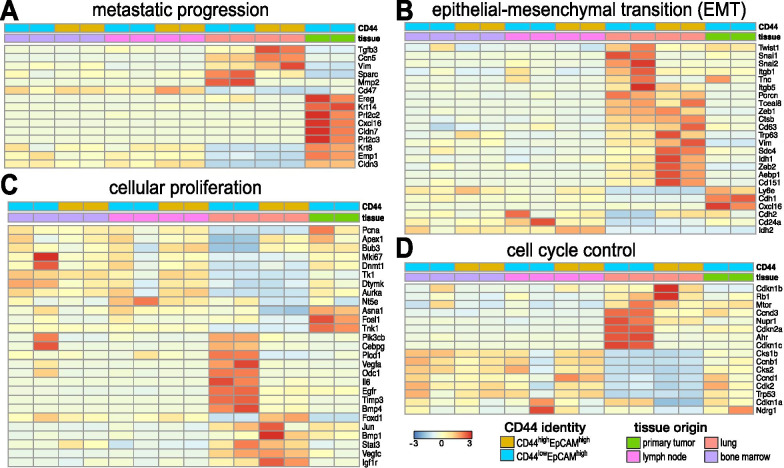
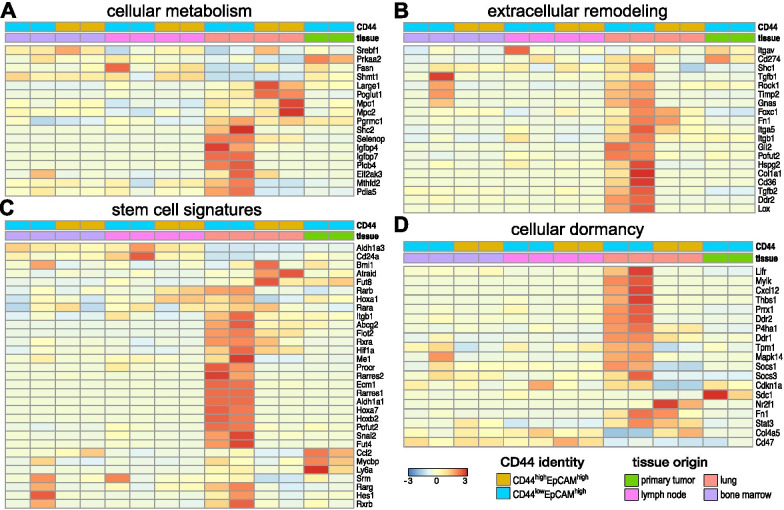
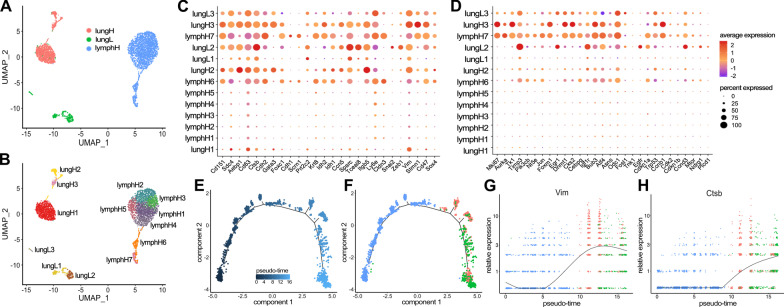
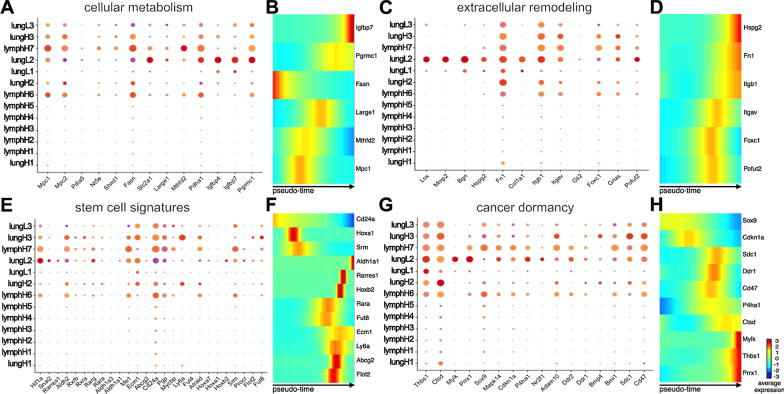
Comparison of RNA sequencing data across all cell populations produced distinct gene clusters. Differential gene expression patterns related to CD44 expression, organ tropism, and immunomodulatory signatures were observed. scRNA-seq identified expression profiles based on tissue-dependent niches and clonal heterogeneity. These cohorts of data were narrowed down to identify subsets of genes with high expression and known metastatic propensity. Dot plot analyses further revealed clusters expressing cancer stem cell and cancer dormancy markers. Changes in relevant genes were investigated across pseudo-time and tissue origin using Monocle2. These data revealed transcriptomes that may contribute to sub-clonal evolution and treatment evasion during cancer progression.
We performed a comprehensive transcriptome analysis of tumor heterogeneity and organ tropism during breast cancer metastasis. These data add to our understanding of metastatic progression and highlight targets for breast cancer treatment. These markers could also be used to image the impact of tumor heterogeneity on metastases.
癌症转移是一个复杂的过程,涉及恶性细胞从原发性肿瘤向远处器官的扩散。从机制层面上理解这一级联反应,可以为该疾病提供关键的新见解,并可能为治疗提供新途径。自发癌症模型的转录组谱分析是一种研究伴随肿瘤细胞扩散的动态变化的有吸引力的方法。然而,这种研究受到所涉及的细胞类型的内在异质性的影响。本研究的目的是使用成熟的 MMTV-PyMT 小鼠模型研究转移性乳腺癌细胞的转录组。
从 10 只雌性 MMTV-PyMT 小鼠中收获器官来源的转移性细胞系。根据 CD44/EpCAM 或 CD44/EpCAM 表面标志物的表达,分离和分选癌细胞。从每个细胞系提取 RNA,并使用 NextSeq 500 Illumina 平台进行测序。比较不同转移性和原发性肿瘤样本中的组织特异性基因。使用 STAR 将reads 映射到小鼠基因组,使用 RSEM 定量基因表达。使用 BioRad 的 ddSeq 平台对选定样本进行单细胞 RNA-seq(scRNA-seq),并使用 Seurat v3.2.3 进行分析。使用 Monocle2 推断伪时间进展。
对所有细胞群体的 RNA 测序数据进行比较,产生了不同的基因簇。观察到与 CD44 表达、器官趋向性和免疫调节特征相关的差异基因表达模式。scRNA-seq 根据组织依赖的生态位和克隆异质性鉴定表达谱。这些数据群集缩小到识别具有高表达和已知转移性倾向的基因子集。点图分析进一步揭示了表达癌症干细胞和癌症休眠标志物的簇。使用 Monocle2 跨伪时间和组织起源研究相关基因的变化。这些数据揭示了可能有助于癌症进展过程中亚克隆进化和治疗逃避的转录组。
我们对乳腺癌转移过程中的肿瘤异质性和器官趋向性进行了全面的转录组分析。这些数据增加了我们对转移性进展的理解,并强调了乳腺癌治疗的靶点。这些标志物也可用于成像肿瘤异质性对转移的影响。