Mora-Jimenez Lucia, Valencia Miguel, Sanchez-Carpintero Rocio, Tønnesen Jan, Fadila Saja, Rubinstein Moran, Gonzalez-Aparicio Manuela, Bunuales Maria, Fernandez-Pierola Eva, Nicolas Maria Jesus, Puerta Elena, Miguelez Cristina, Minguez Paula Gimenez, Lumbreras Sara, Gonzalez-Aseguinolaza Gloria, Ricobaraza Ana, Hernandez-Alcoceba Ruben
Gene Therapy and Regulation of Gene Expression Program, CIMA, University of Navarra, IdiSNA, Navarra Institute for Health Research, Pamplona, Spain.
Neuroscience Program, CIMA, University of Navarra, IdiSNA, Navarra Institute for Health Research, Pamplona, Spain.
Mol Ther Nucleic Acids. 2021 Aug 19;25:585-602. doi: 10.1016/j.omtn.2021.08.003. eCollection 2021 Sep 3.
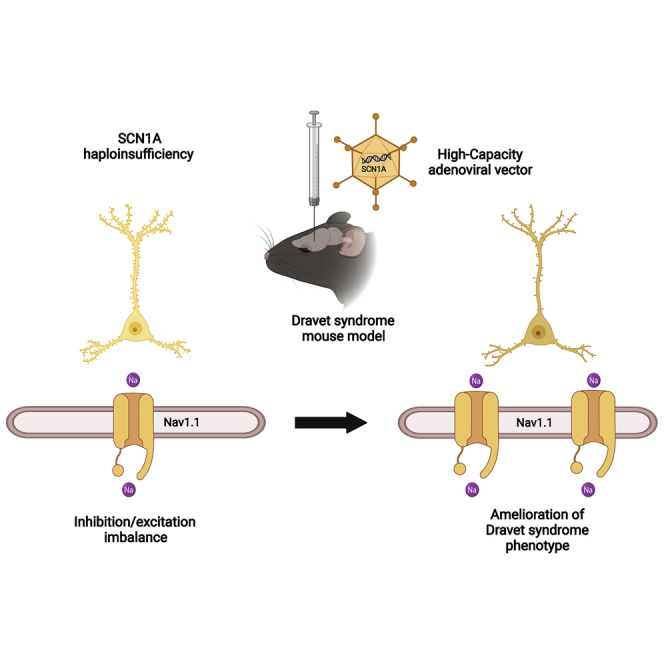
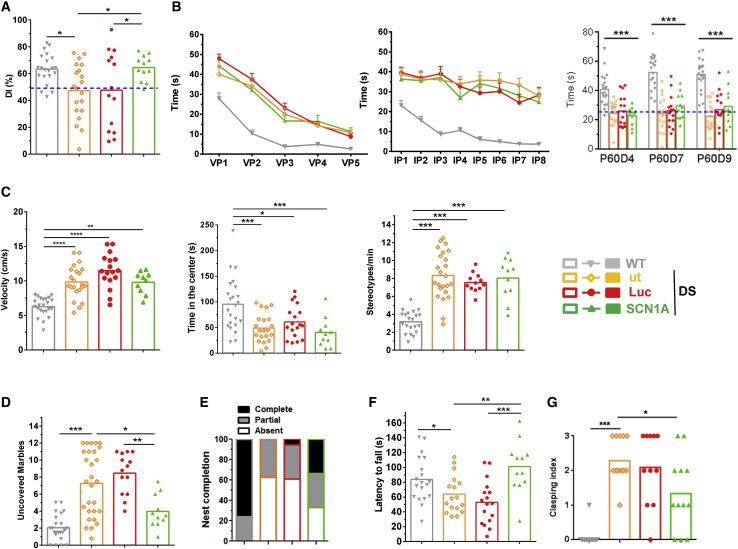
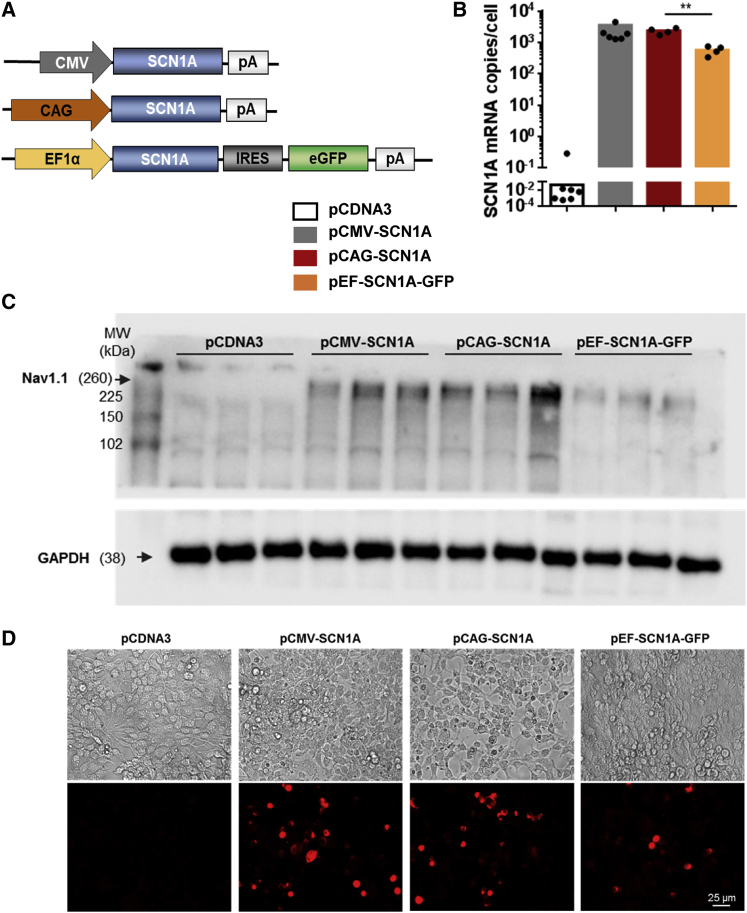
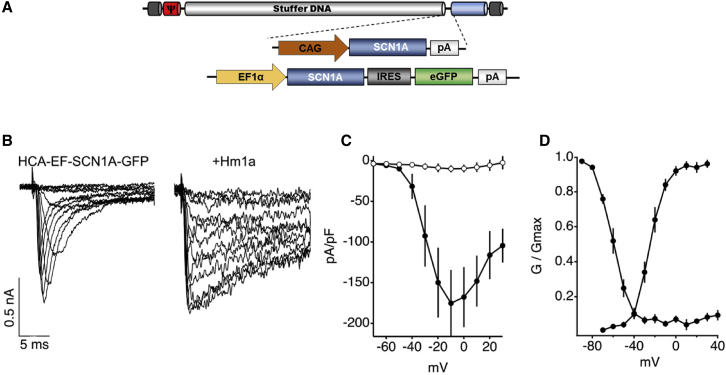
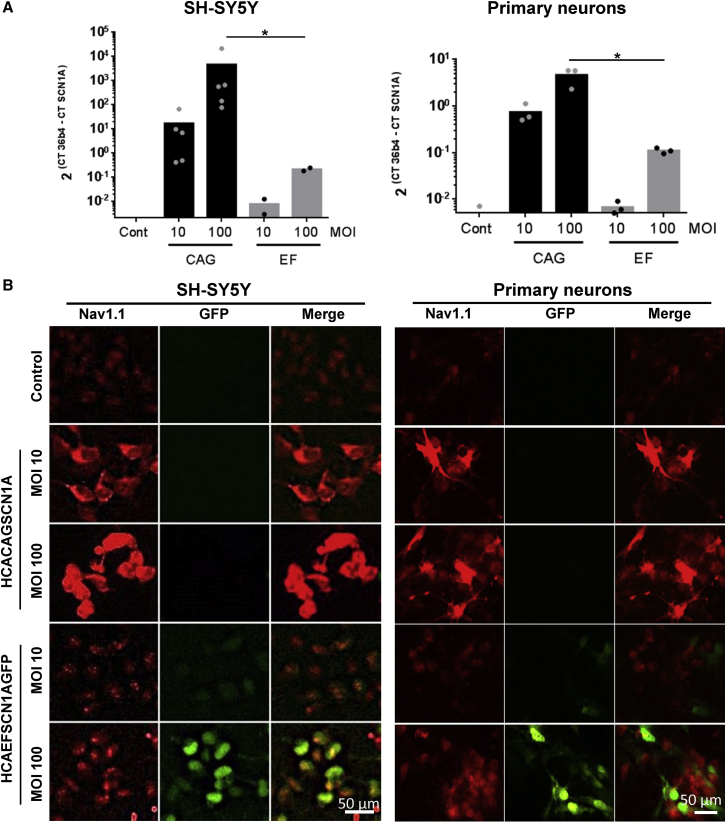
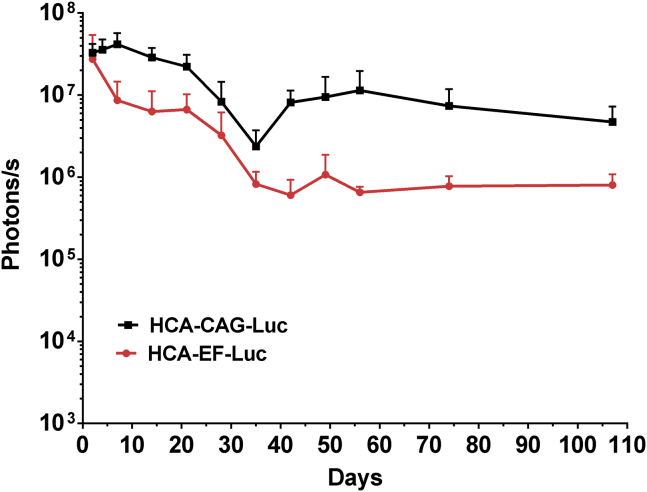
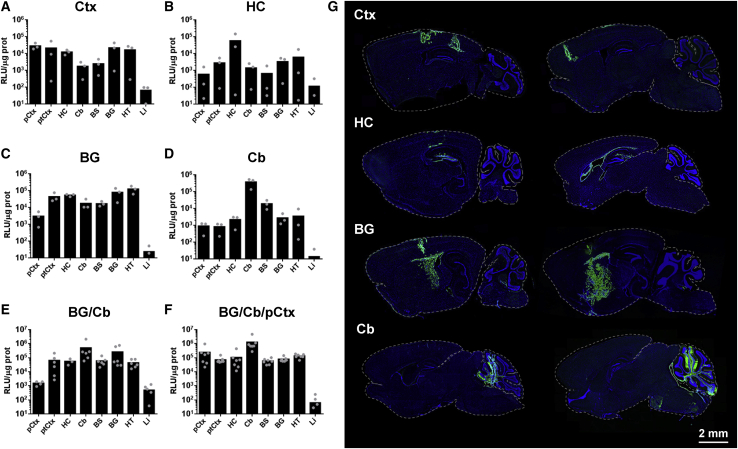
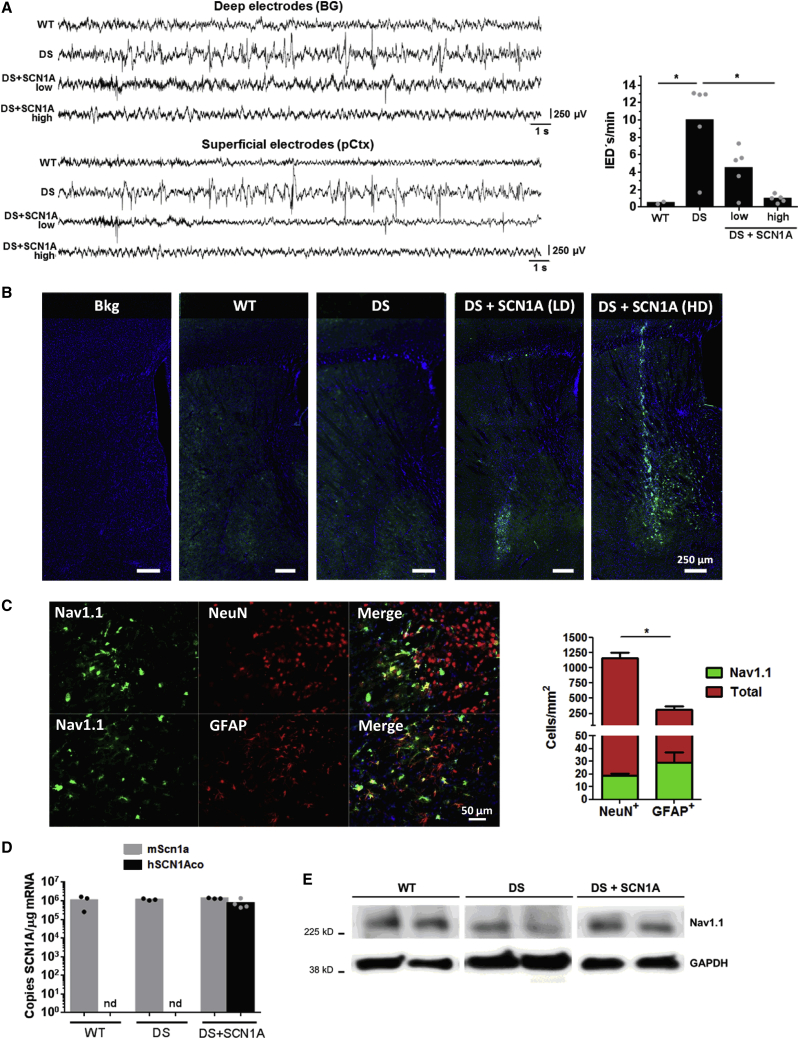
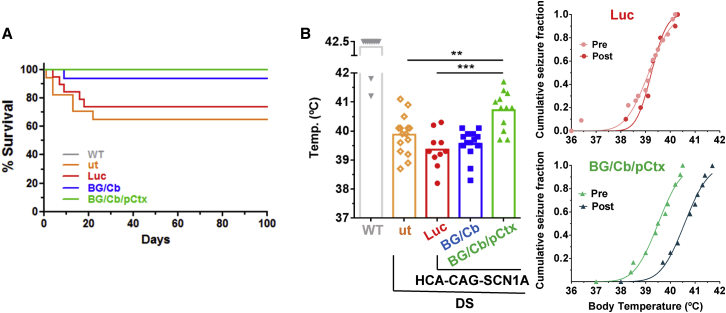
Dravet syndrome is a genetic encephalopathy characterized by severe epilepsy combined with motor, cognitive, and behavioral abnormalities. Current antiepileptic drugs achieve only partial control of seizures and provide little benefit on the patient's neurological development. In >80% of cases, the disease is caused by haploinsufficiency of the gene, which encodes the alpha subunit of the Nav1.1 voltage-gated sodium channel. Novel therapies aim to restore expression in order to address all disease manifestations. We provide evidence that a high-capacity adenoviral vector harboring the 6-kb cDNA is feasible and able to express functional Nav1.1 in neurons. , the best biodistribution was observed after intracerebral injection in basal ganglia, cerebellum, and prefrontal cortex. A1783V knockin mice received the vector at 5 weeks of age, when most neurological alterations were present. Animals were protected from sudden death, and the epileptic phenotype was attenuated. Improvement of motor performance and interaction with the environment was observed. In contrast, hyperactivity persisted, and the impact on cognitive tests was variable (success in novel object recognition and failure in Morris water maze tests). These results provide proof of concept for gene supplementation in Dravet syndrome and indicate new directions for improvement.
德拉韦综合征是一种遗传性脑病,其特征为严重癫痫合并运动、认知和行为异常。目前的抗癫痫药物仅能部分控制癫痫发作,对患者的神经发育益处甚微。在超过80%的病例中,该疾病由编码Nav1.1电压门控钠通道α亚基的基因单倍剂量不足引起。新型疗法旨在恢复该基因的表达,以解决所有疾病表现。我们提供的证据表明,携带6 kb该基因cDNA的高容量腺病毒载体是可行的,并且能够在神经元中表达功能性Nav1.1。在基底神经节、小脑和前额叶皮质进行脑内注射后,观察到了最佳的生物分布。A1783V基因敲入小鼠在5周龄时接受该载体,此时大多数神经学改变已经出现。动物免受猝死,癫痫表型得到缓解。观察到运动表现有所改善,与环境的互动也有所改善。相比之下,多动症状持续存在,对认知测试的影响则各不相同(在新物体识别测试中成功,在莫里斯水迷宫测试中失败)。这些结果为德拉韦综合征的基因补充提供了概念验证,并指明了改进的新方向。