Department of Computer Science, Texas A&M University, College Station, TX, United States of America.
Laboratory of Host-Pathogen Biology, The Rockefeller University, New York, NY, United States of America.
PLoS One. 2021 Oct 1;16(10):e0257911. doi: 10.1371/journal.pone.0257911. eCollection 2021.
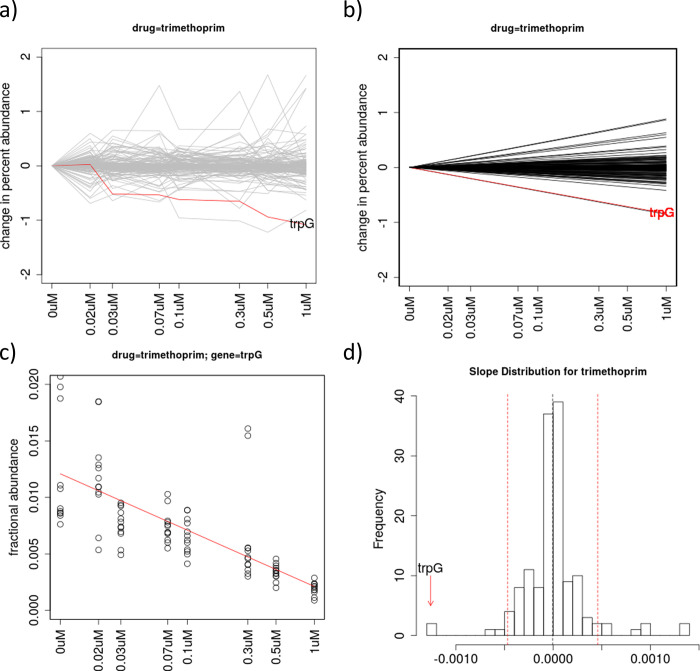
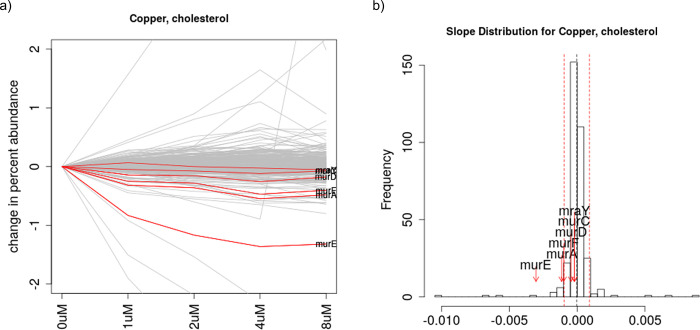
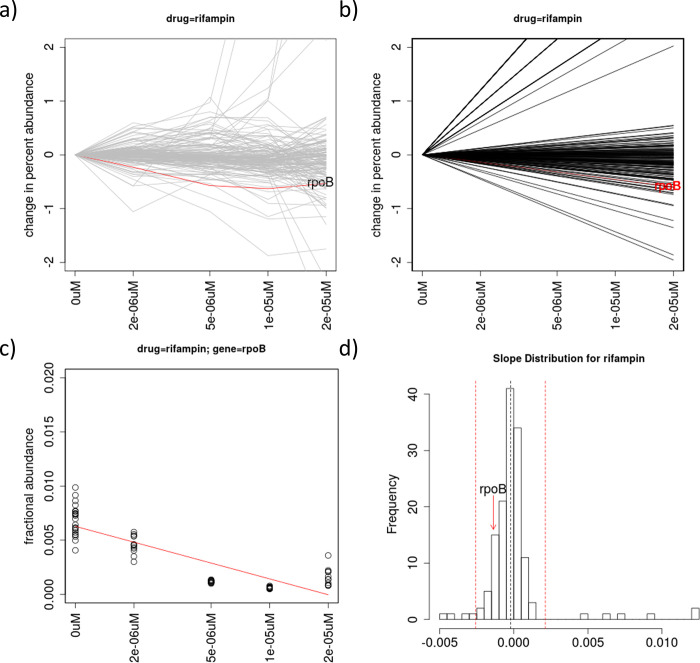
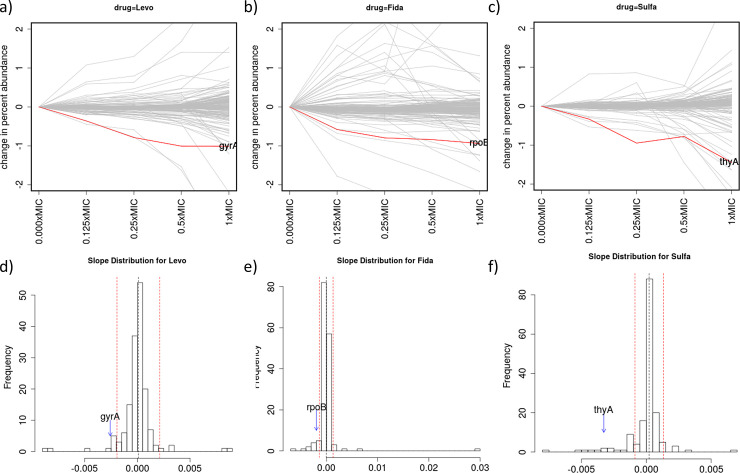
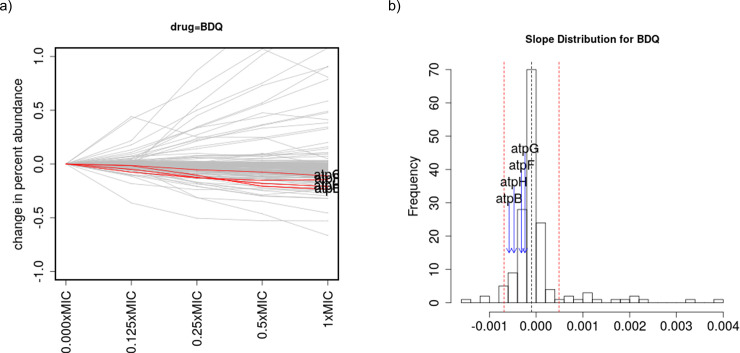
Chemical-genetics (C-G) experiments can be used to identify interactions between inhibitory compounds and bacterial genes, potentially revealing the targets of drugs, or other functionally interacting genes and pathways. C-G experiments involve constructing a library of hypomorphic strains with essential genes that can be knocked-down, treating it with an inhibitory compound, and using high-throughput sequencing to quantify changes in relative abundance of individual mutants. The hypothesis is that, if the target of a drug or other genes in the same pathway are present in the library, such genes will display an excessive fitness defect due to the synergy between the dual stresses of protein depletion and antibiotic exposure. While assays at a single drug concentration are susceptible to noise and can yield false-positive interactions, improved detection can be achieved by requiring that the synergy between gene and drug be concentration-dependent. We present a novel statistical method based on Linear Mixed Models, called CGA-LMM, for analyzing C-G data. The approach is designed to capture the dependence of the abundance of each gene in the hypomorph library on increasing concentrations of drug through slope coefficients. To determine which genes represent candidate interactions, CGA-LMM uses a conservative population-based approach in which genes with negative slopes are considered significant only if they are outliers with respect to the rest of the population (assuming that most genes in the library do not interact with a given inhibitor). We applied the method to analyze 3 independent hypomorph libraries of M. tuberculosis for interactions with antibiotics with anti-tubercular activity, and we identify known target genes or expected interactions for 7 out of 9 drugs where relevant interacting genes are known.
化学遗传学(C-G)实验可用于鉴定抑制性化合物与细菌基因之间的相互作用,从而可能揭示药物的靶标或其他具有功能相互作用的基因和途径。C-G 实验涉及构建一个具有必需基因的弱表型菌株文库,这些基因可以被敲低,用抑制性化合物处理,然后使用高通量测序来定量个体突变体相对丰度的变化。假设如果药物的靶标或同一途径中的其他基因存在于文库中,由于蛋白耗竭和抗生素暴露的双重压力协同作用,这些基因将表现出过度的适应性缺陷。虽然在单一药物浓度下进行的测定容易受到噪声的影响,并可能产生假阳性相互作用,但通过要求基因与药物之间的协同作用与浓度相关,可以提高检测效果。我们提出了一种基于线性混合模型的新统计方法,称为 CGA-LMM,用于分析 C-G 数据。该方法旨在通过斜率系数捕获弱表型文库中每个基因的丰度对药物浓度增加的依赖性。为了确定哪些基因代表候选相互作用,CGA-LMM 使用了一种保守的基于群体的方法,其中具有负斜率的基因只有在相对于群体其他部分(假设文库中的大多数基因与给定抑制剂不相互作用)是异常值时才被认为是显著的。我们应用该方法分析了 3 个独立的结核分枝杆菌弱表型文库与具有抗结核活性的抗生素的相互作用,我们确定了 9 种药物中的 7 种已知靶标基因或预期相互作用,其中 7 种药物是已知的相关相互作用基因。