Bornbusch Sally L, Harris Rachel L, Grebe Nicholas M, Roche Kimberly, Dimac-Stohl Kristin, Drea Christine M
Department of Evolutionary Anthropology, Duke University, Durham, USA.
Program in Computational Biology & Bioinformatics, Duke University, Durham, USA.
Anim Microbiome. 2021 Oct 1;3(1):65. doi: 10.1186/s42523-021-00126-z.
Antibiotics alter the diversity, structure, and dynamics of host-associated microbial consortia, including via development of antibiotic resistance; however, patterns of recovery from microbial imbalances and methods to mitigate associated negative effects remain poorly understood, particularly outside of human-clinical and model-rodent studies that focus on outcome over process. To improve conceptual understanding of host-microbe symbiosis in more naturalistic contexts, we applied an ecological framework to a non-traditional, strepsirrhine primate model via long-term, multi-faceted study of microbial community structure before, during, and following two experimental manipulations. Specifically, we administered a broad-spectrum antibiotic, either alone or with subsequent fecal transfaunation, to healthy, male ring-tailed lemurs (Lemur catta), then used 16S rRNA and shotgun metagenomic sequencing to longitudinally track the diversity, composition, associations, and resistomes of their gut microbiota both within and across baseline, treatment, and recovery phases.
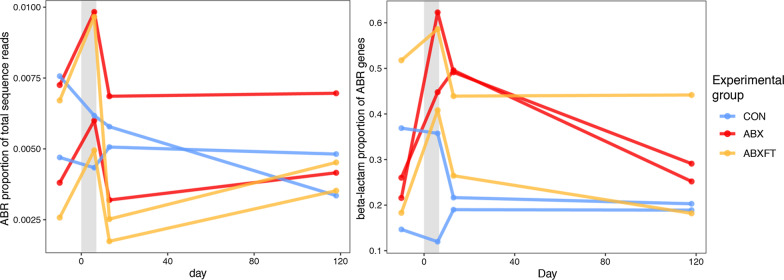
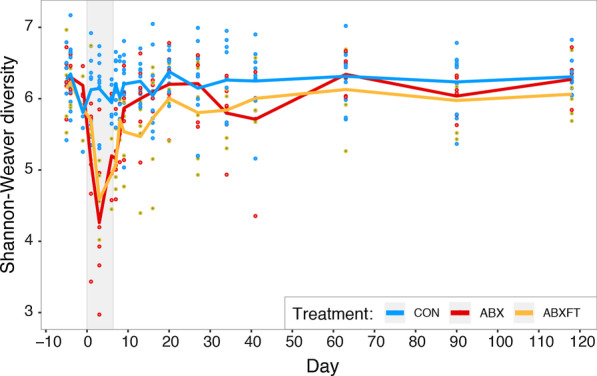
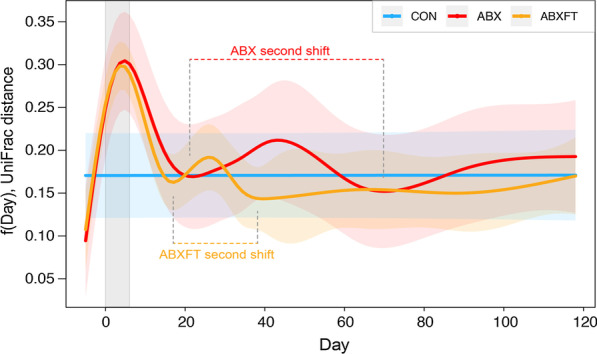
Antibiotic treatment resulted in a drastic decline in microbial diversity and a dramatic alteration in community composition. Whereas microbial diversity recovered rapidly regardless of experimental group, patterns of microbial community composition reflected long-term instability following treatment with antibiotics alone, a pattern that was attenuated by fecal transfaunation. Covariation analysis revealed that certain taxa dominated bacterial associations, representing potential keystone species in lemur gut microbiota. Antibiotic resistance genes, which were universally present, including in lemurs that had never been administered antibiotics, varied across individuals and treatment groups.
Long-term, integrated study post antibiotic-induced microbial imbalance revealed differential, metric-dependent evidence of recovery, with beneficial effects of fecal transfaunation on recovering community composition, and potentially negative consequences to lemur resistomes. Beyond providing new perspectives on the dynamics that govern host-associated communities, particularly in the Anthropocene era, our holistic study in an endangered species is a first step in addressing the recent, interdisciplinary calls for greater integration of microbiome science into animal care and conservation.
抗生素会改变宿主相关微生物群落的多样性、结构和动态,包括通过产生抗生素耐药性来实现;然而,从微生物失衡中恢复的模式以及减轻相关负面影响的方法仍知之甚少,尤其是在关注结果而非过程的人类临床和啮齿动物模型研究之外。为了在更自然的环境中增进对宿主 - 微生物共生关系的概念理解,我们通过对两个实验操作之前、期间和之后的微生物群落结构进行长期、多方面的研究,将一个生态框架应用于一个非传统的狐猴灵长类动物模型。具体而言,我们对健康的雄性环尾狐猴(Lemur catta)单独或在随后进行粪便微生物移植的情况下给予广谱抗生素,然后使用16S rRNA和鸟枪法宏基因组测序在基线、治疗和恢复阶段纵向追踪其肠道微生物群的多样性、组成、关联和耐药组。
抗生素治疗导致微生物多样性急剧下降,群落组成发生显著改变。尽管无论实验组如何,微生物多样性都迅速恢复,但微生物群落组成模式反映出单独使用抗生素治疗后长期的不稳定性,而粪便微生物移植减弱了这种模式。共变分析表明,某些分类群主导细菌关联,代表了狐猴肠道微生物群中潜在的关键物种。抗生素耐药基因普遍存在,包括在从未接受过抗生素治疗的狐猴中,且在个体和治疗组之间存在差异。
抗生素诱导的微生物失衡后的长期综合研究揭示了不同的、依赖指标的恢复证据,粪便微生物移植对恢复群落组成有有益影响,且可能对狐猴耐药组产生负面后果。除了为控制宿主相关群落的动态提供新视角外,尤其是在人类世时代,我们在濒危物种中的整体研究是应对近期跨学科呼吁将微生物组科学更全面地整合到动物护理和保护中的第一步。