Sorbonne Université, CNRS, Institut de Biologie Paris Seine, Biologie Computationnelle et Quantitative LCQB, F-75005, Paris, France.
Laboratoire de Physique de l'Ecole Normale Supérieure, ENS, Université PSL, CNRS, Sorbonne Université, Université de Paris, F-75005, Paris, France.
Nat Commun. 2021 Oct 4;12(1):5800. doi: 10.1038/s41467-021-25756-4.
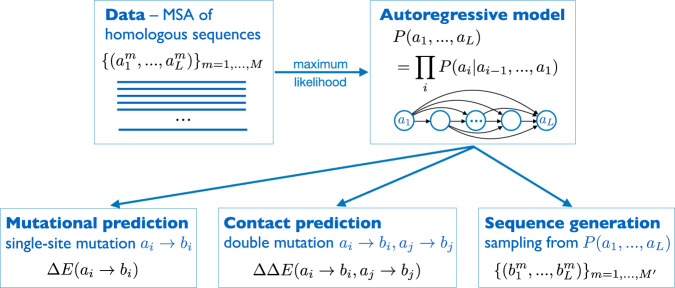
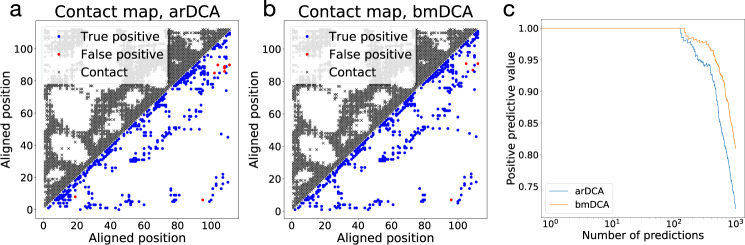
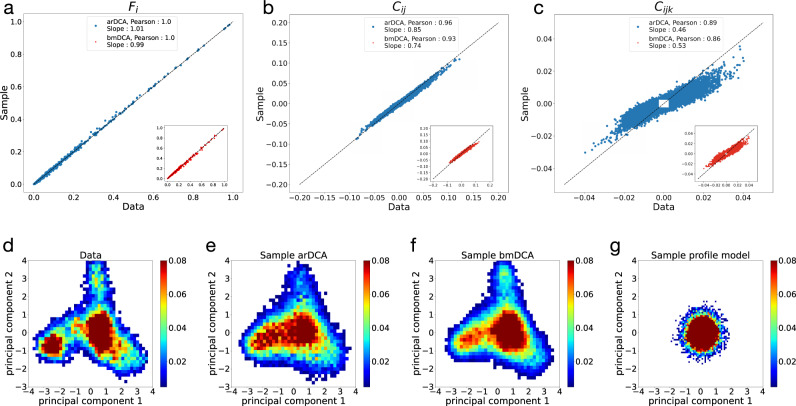
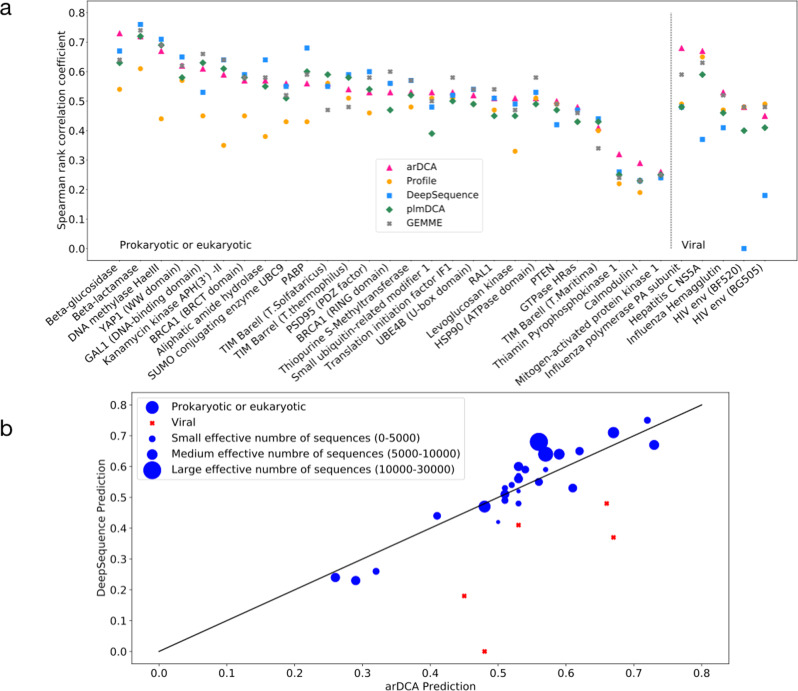
Generative models emerge as promising candidates for novel sequence-data driven approaches to protein design, and for the extraction of structural and functional information about proteins deeply hidden in rapidly growing sequence databases. Here we propose simple autoregressive models as highly accurate but computationally efficient generative sequence models. We show that they perform similarly to existing approaches based on Boltzmann machines or deep generative models, but at a substantially lower computational cost (by a factor between 10 and 10). Furthermore, the simple structure of our models has distinctive mathematical advantages, which translate into an improved applicability in sequence generation and evaluation. Within these models, we can easily estimate both the probability of a given sequence, and, using the model's entropy, the size of the functional sequence space related to a specific protein family. In the example of response regulators, we find a huge number of ca. 10 possible sequences, which nevertheless constitute only the astronomically small fraction 10 of all amino-acid sequences of the same length. These findings illustrate the potential and the difficulty in exploring sequence space via generative sequence models.
生成模型是一种很有前途的候选方法,可用于蛋白质设计中的新型序列数据驱动方法,也可用于从快速增长的序列数据库中提取蛋白质的结构和功能信息。在这里,我们提出了简单的自回归模型,作为高度准确但计算效率高的生成序列模型。我们表明,它们与基于玻尔兹曼机或深度生成模型的现有方法表现相似,但计算成本要低得多(低 10 到 10 倍)。此外,我们模型的简单结构具有独特的数学优势,这转化为在序列生成和评估中的更好适用性。在这些模型中,我们可以轻松估计给定序列的概率,并且可以使用模型的熵来评估与特定蛋白质家族相关的功能序列空间的大小。在响应调节剂的示例中,我们发现了大量大约 10 种可能的序列,但它们仅占相同长度的所有氨基酸序列的天文数字小部分 10。这些发现说明了通过生成序列模型探索序列空间的潜力和困难。