Department of Biochemistry, University of Washington, Seattle, WA 98105.
Institute for Protein Design, University of Washington, Seattle, WA 98105.
Proc Natl Acad Sci U S A. 2021 Mar 16;118(11). doi: 10.1073/pnas.2017228118.
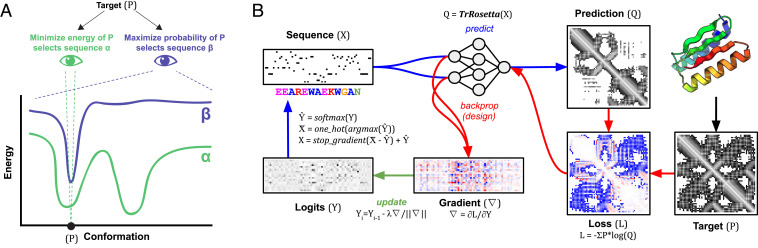
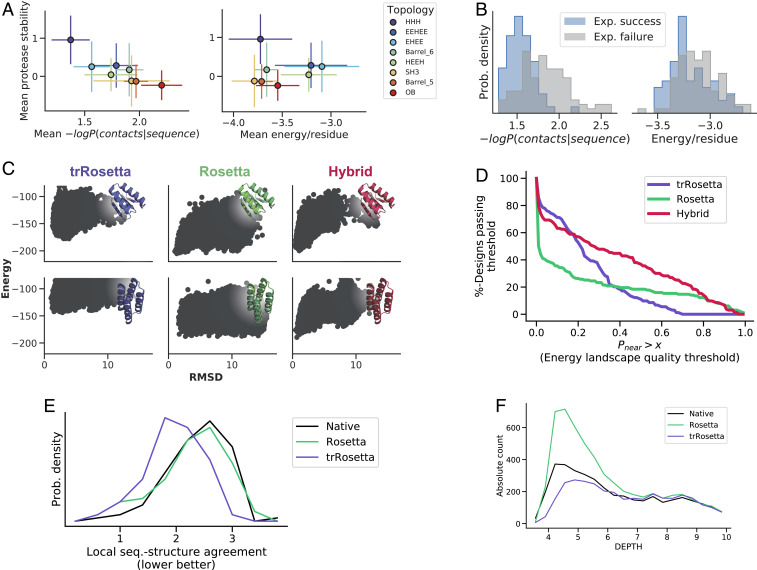
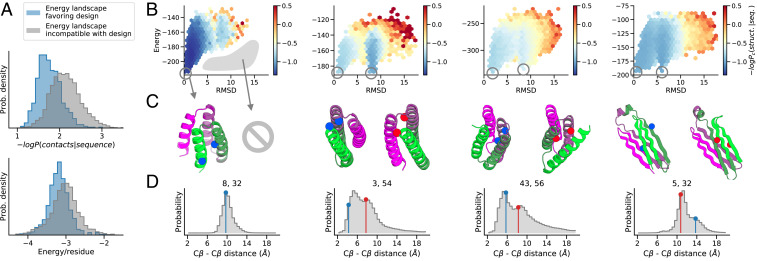
The protein design problem is to identify an amino acid sequence that folds to a desired structure. Given Anfinsen's thermodynamic hypothesis of folding, this can be recast as finding an amino acid sequence for which the desired structure is the lowest energy state. As this calculation involves not only all possible amino acid sequences but also, all possible structures, most current approaches focus instead on the more tractable problem of finding the lowest-energy amino acid sequence for the desired structure, often checking by protein structure prediction in a second step that the desired structure is indeed the lowest-energy conformation for the designed sequence, and typically discarding a large fraction of designed sequences for which this is not the case. Here, we show that by backpropagating gradients through the transform-restrained Rosetta (trRosetta) structure prediction network from the desired structure to the input amino acid sequence, we can directly optimize over all possible amino acid sequences and all possible structures in a single calculation. We find that trRosetta calculations, which consider the full conformational landscape, can be more effective than Rosetta single-point energy estimations in predicting folding and stability of de novo designed proteins. We compare sequence design by conformational landscape optimization with the standard energy-based sequence design methodology in Rosetta and show that the former can result in energy landscapes with fewer alternative energy minima. We show further that more funneled energy landscapes can be designed by combining the strengths of the two approaches: the low-resolution trRosetta model serves to disfavor alternative states, and the high-resolution Rosetta model serves to create a deep energy minimum at the design target structure.
蛋白质设计问题是确定一个氨基酸序列,使其折叠成所需的结构。根据 Anfinsen 的折叠热力学假设,这可以重新表述为找到一个氨基酸序列,其中所需的结构是最低能量状态。由于这个计算不仅涉及所有可能的氨基酸序列,还涉及所有可能的结构,因此目前大多数方法都集中在寻找所需结构的最低能量氨基酸序列的更可处理的问题上,通常在第二步通过蛋白质结构预测来检查所需的结构是否确实是设计序列的最低能量构象,并且通常会丢弃很大一部分不满足此条件的设计序列。在这里,我们表明,通过从所需结构向后传播梯度到输入的氨基酸序列,我们可以在单个计算中直接优化所有可能的氨基酸序列和所有可能的结构。我们发现,考虑到完整构象景观的 trRosetta 结构预测网络计算可以比 Rosetta 单点能量估计更有效地预测从头设计的蛋白质的折叠和稳定性。我们将构象景观优化的序列设计与 Rosetta 中的标准基于能量的序列设计方法进行比较,并表明前者可以产生具有更少替代能量最小值的能量景观。我们进一步表明,通过结合两种方法的优势,可以设计出更具漏斗形的能量景观:低分辨率的 trRosetta 模型有助于不利替代状态,而高分辨率的 Rosetta 模型有助于在设计目标结构处创建深的能量最低点。