Patel Chirag N, Jani Siddhi P, Jaiswal Dharmesh G, Kumar Sivakumar Prasanth, Mangukia Naman, Parmar Robin M, Rawal Rakesh M, Pandya Himanshu A
Department of Botany, Bioinformatics, and Climate Change Impacts Management, School of Sciences, Gujarat University, Ahmedabad, 380009, India.
BioInnovations, Bhayander (West), Mumbai, 401101, India.
Sci Rep. 2021 Oct 13;11(1):20295. doi: 10.1038/s41598-021-99165-4.
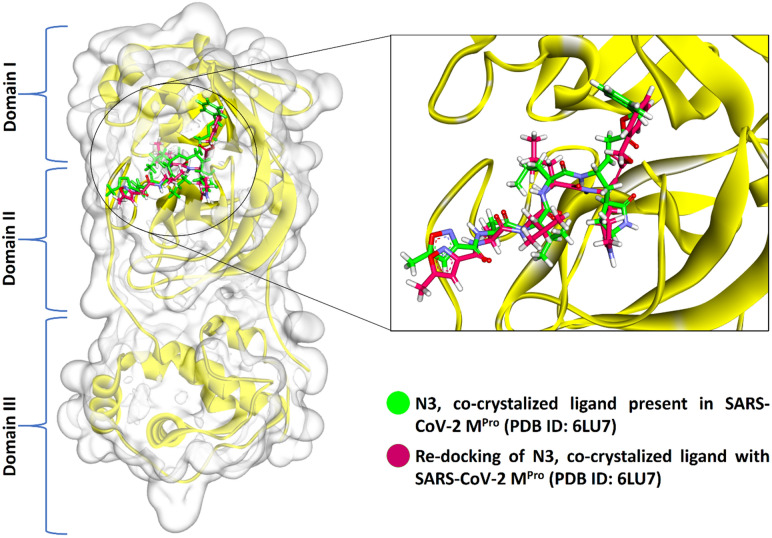
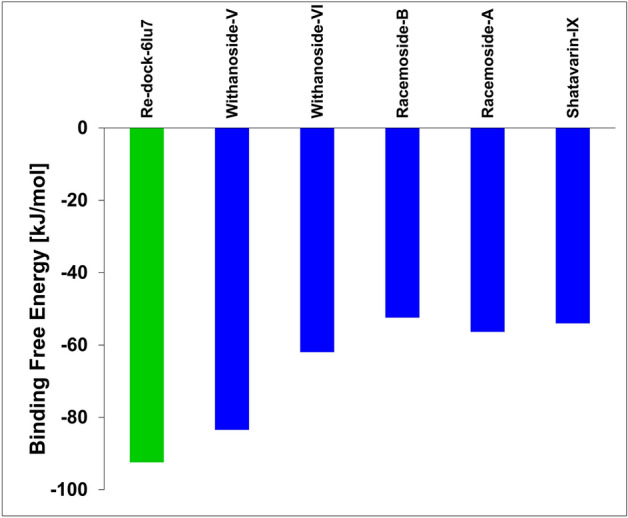
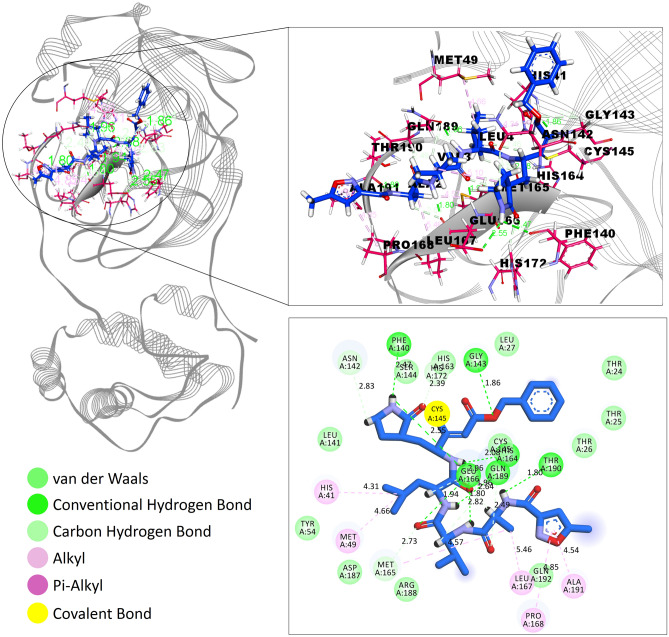
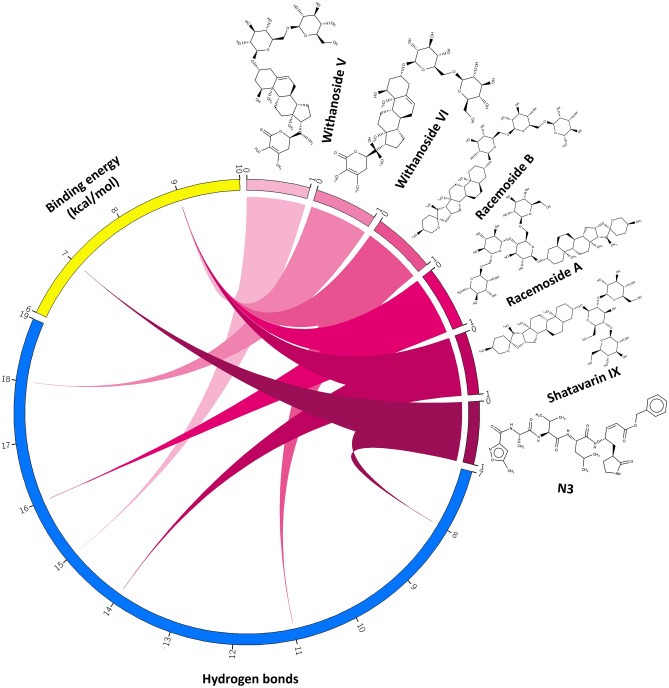
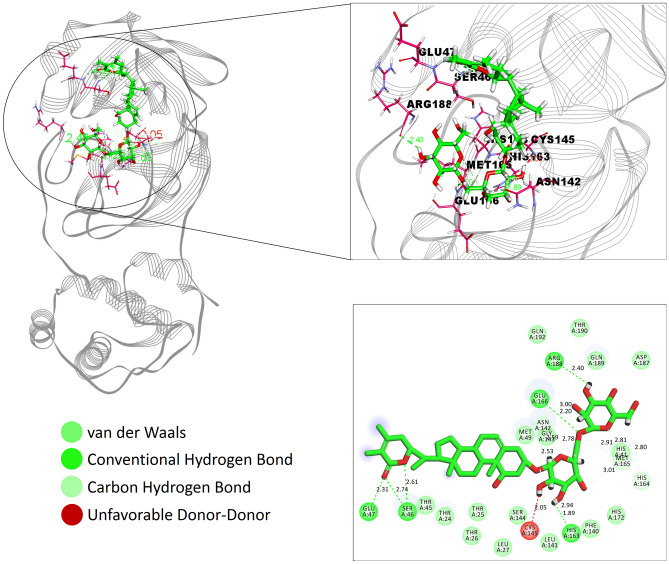
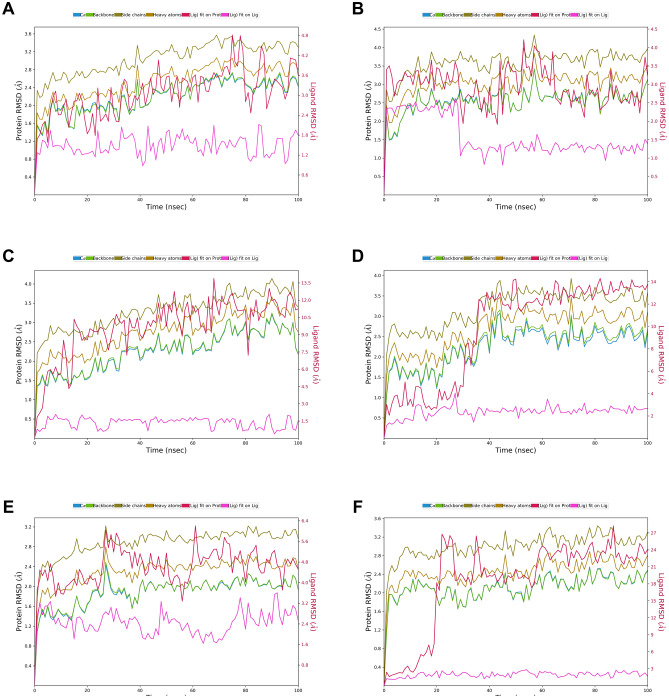
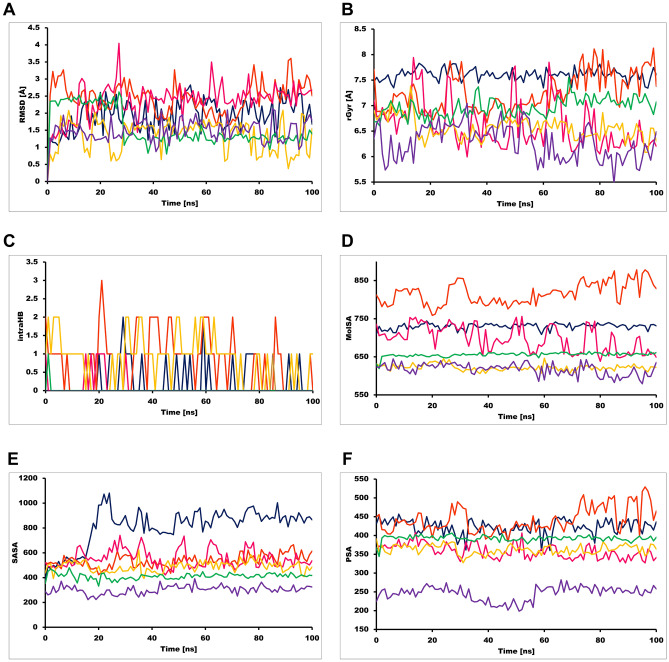
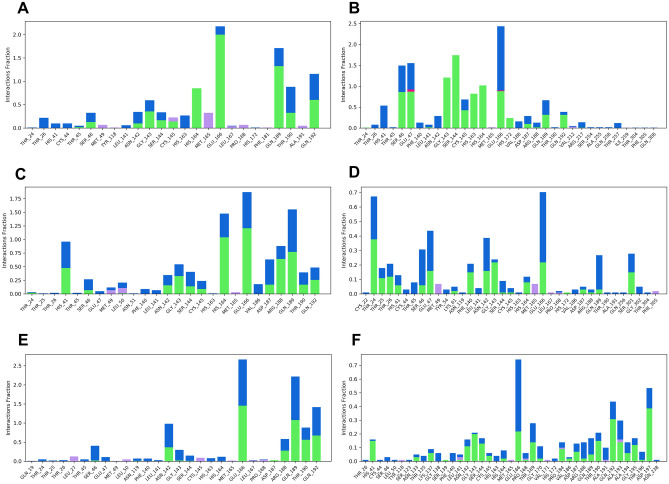
Novel SARS-CoV-2, an etiological factor of Coronavirus disease 2019 (COVID-19), poses a great challenge to the public health care system. Among other druggable targets of SARS-Cov-2, the main protease (M) is regarded as a prominent enzyme target for drug developments owing to its crucial role in virus replication and transcription. We pursued a computational investigation to identify M inhibitors from a compiled library of natural compounds with proven antiviral activities using a hierarchical workflow of molecular docking, ADMET assessment, dynamic simulations and binding free-energy calculations. Five natural compounds, Withanosides V and VI, Racemosides A and B, and Shatavarin IX, obtained better binding affinity and attained stable interactions with M key pocket residues. These intermolecular key interactions were also retained profoundly in the simulation trajectory of 100 ns time scale indicating tight receptor binding. Free energy calculations prioritized Withanosides V and VI as the top candidates that can act as effective SARS-CoV-2 M inhibitors.
新型严重急性呼吸综合征冠状病毒2(SARS-CoV-2)是2019冠状病毒病(COVID-19)的病原体,给公共卫生保健系统带来了巨大挑战。在SARS-CoV-2的其他可成药靶点中,主要蛋白酶(M)因其在病毒复制和转录中的关键作用,被视为药物开发的重要酶靶点。我们进行了一项计算研究,使用分子对接、ADMET评估、动力学模拟和结合自由能计算的分层工作流程,从一个已编译的具有抗病毒活性的天然化合物库中识别M抑制剂。五种天然化合物,即睡茄苷V和VI、总状土木香苷A和B以及印度人参苷IX,获得了更好的结合亲和力,并与M关键口袋残基形成了稳定的相互作用。这些分子间的关键相互作用在100纳秒时间尺度的模拟轨迹中也得到了深刻保留,表明受体结合紧密。自由能计算将睡茄苷V和VI列为可作为有效SARS-CoV-2 M抑制剂的首选候选物。