Shen Chao, Hu Xueping, Gao Junbo, Zhang Xujun, Zhong Haiyang, Wang Zhe, Xu Lei, Kang Yu, Cao Dongsheng, Hou Tingjun
Innovation Institute for Artificial Intelligence in Medicine of Zhejiang University, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang, 310058, People's Republic of China.
State Key Lab of CAD&CG, Zhejiang University, Hangzhou, Zhejiang, 310058, People's Republic of China.
J Cheminform. 2021 Oct 16;13(1):81. doi: 10.1186/s13321-021-00560-w.
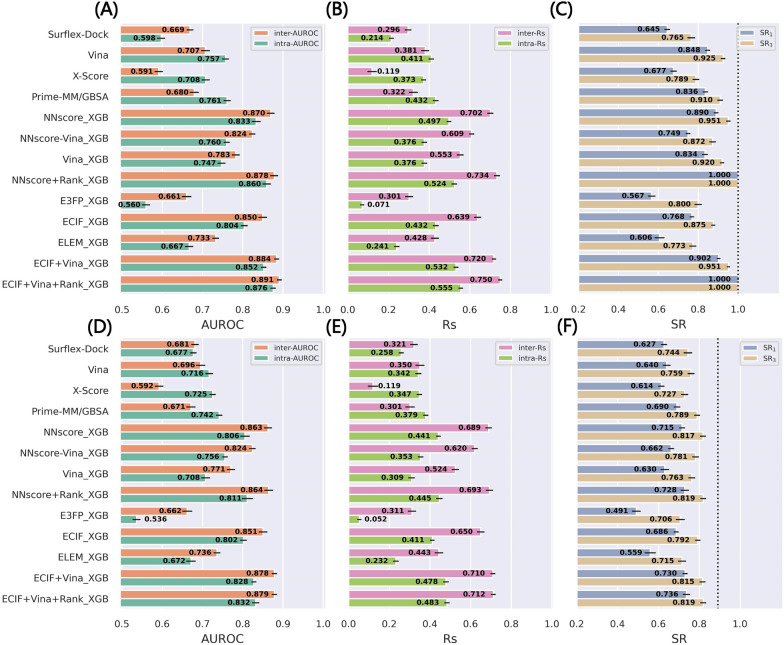
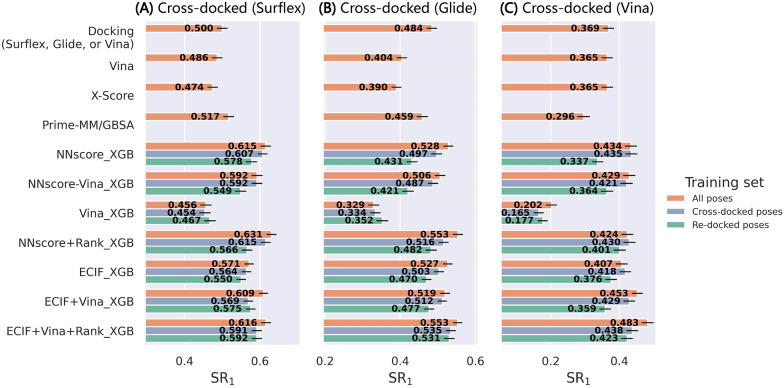
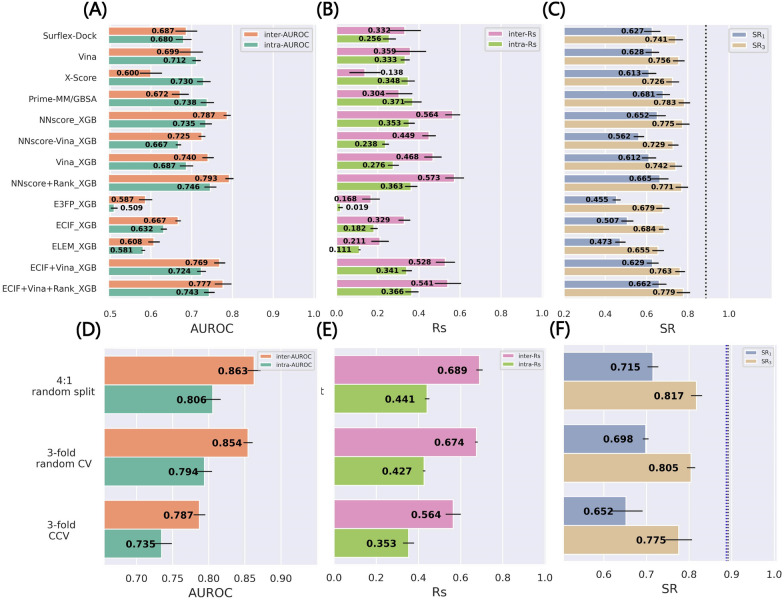
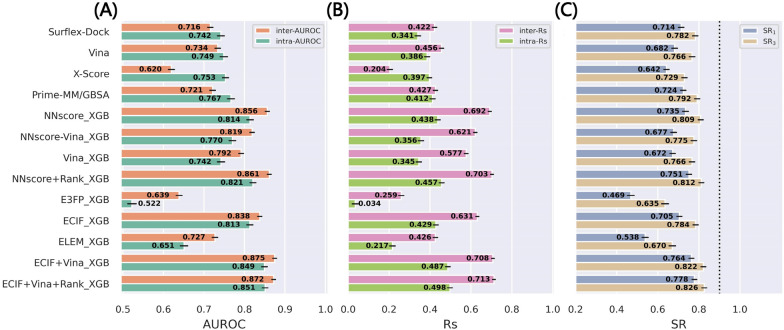
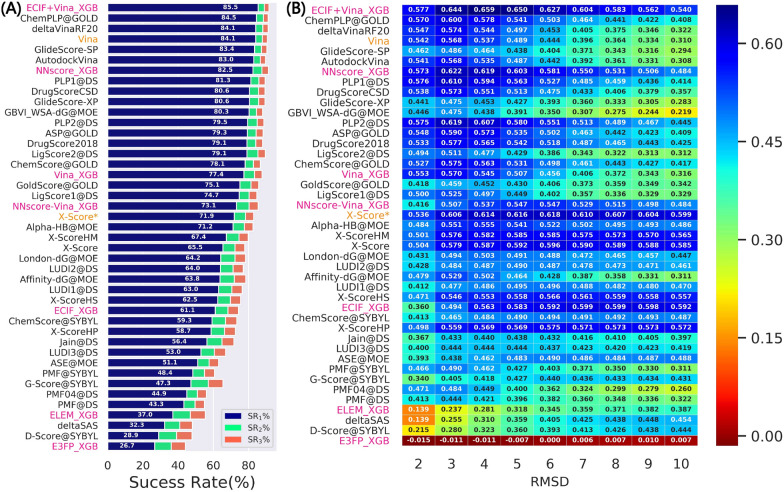
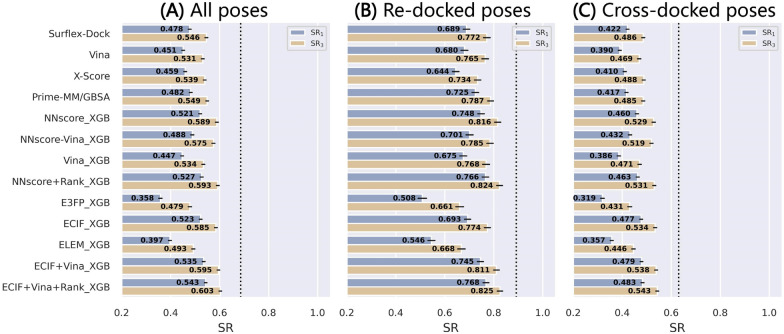
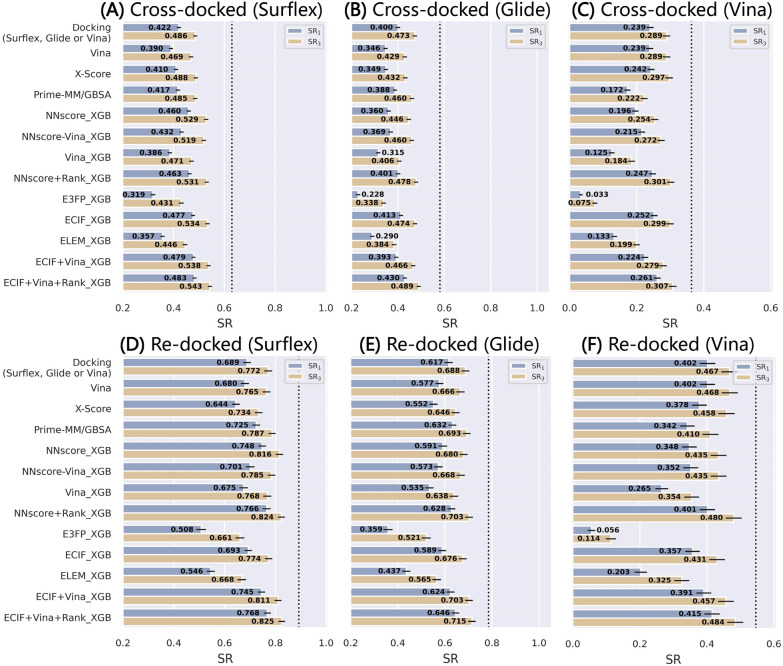
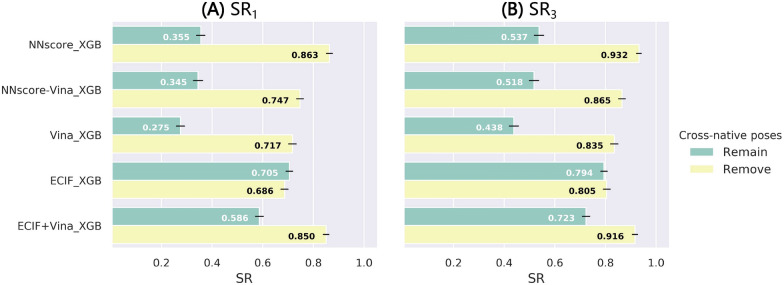
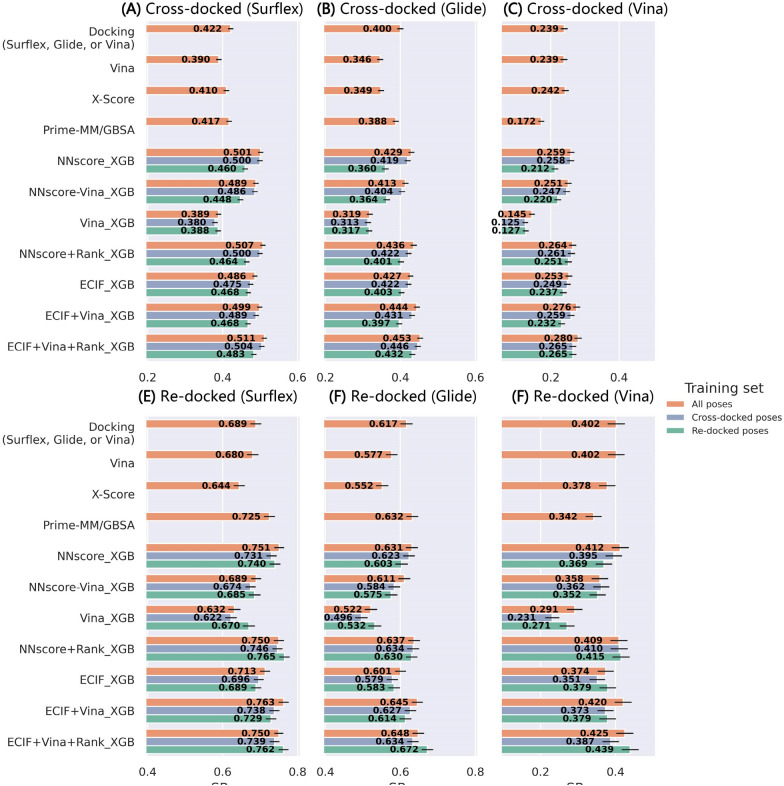
Structure-based drug design depends on the detailed knowledge of the three-dimensional (3D) structures of protein-ligand binding complexes, but accurate prediction of ligand-binding poses is still a major challenge for molecular docking due to deficiency of scoring functions (SFs) and ignorance of protein flexibility upon ligand binding. In this study, based on a cross-docking dataset dedicatedly constructed from the PDBbind database, we developed several XGBoost-trained classifiers to discriminate the near-native binding poses from decoys, and systematically assessed their performance with/without the involvement of the cross-docked poses in the training/test sets. The calculation results illustrate that using Extended Connectivity Interaction Features (ECIF), Vina energy terms and docking pose ranks as the features can achieve the best performance, according to the validation through the random splitting or refined-core splitting and the testing on the re-docked or cross-docked poses. Besides, it is found that, despite the significant decrease of the performance for the threefold clustered cross-validation, the inclusion of the Vina energy terms can effectively ensure the lower limit of the performance of the models and thus improve their generalization capability. Furthermore, our calculation results also highlight the importance of the incorporation of the cross-docked poses into the training of the SFs with wide application domain and high robustness for binding pose prediction. The source code and the newly-developed cross-docking datasets can be freely available at https://github.com/sc8668/ml_pose_prediction and https://zenodo.org/record/5525936 , respectively, under an open-source license. We believe that our study may provide valuable guidance for the development and assessment of new machine learning-based SFs (MLSFs) for the predictions of protein-ligand binding poses.
基于结构的药物设计依赖于蛋白质-配体结合复合物三维(3D)结构的详细知识,但由于评分函数(SFs)的不足以及在配体结合时对蛋白质灵活性的忽视,准确预测配体结合姿势仍然是分子对接的一项重大挑战。在本研究中,基于专门从PDBbind数据库构建的交叉对接数据集,我们开发了几个经XGBoost训练的分类器,以区分近天然结合姿势与诱饵,并在训练/测试集中有/无交叉对接姿势参与的情况下系统地评估了它们的性能。计算结果表明,根据通过随机拆分或精炼核心拆分进行的验证以及对重新对接或交叉对接姿势的测试,使用扩展连接相互作用特征(ECIF)、Vina能量项和对接姿势排名作为特征可以实现最佳性能。此外,还发现,尽管三重聚类交叉验证的性能显著下降,但包含Vina能量项可以有效地确保模型性能的下限,从而提高其泛化能力。此外,我们的计算结果还突出了将交叉对接姿势纳入具有广泛应用领域和高结合姿势预测稳健性的SFs训练中的重要性。源代码和新开发的交叉对接数据集分别可在开源许可下从https://github.com/sc8668/ml_pose_prediction和https://zenodo.org/record/5525936免费获取。我们相信,我们的研究可能为开发和评估用于预测蛋白质-配体结合姿势的新型基于机器学习的SFs(MLSFs)提供有价值的指导。