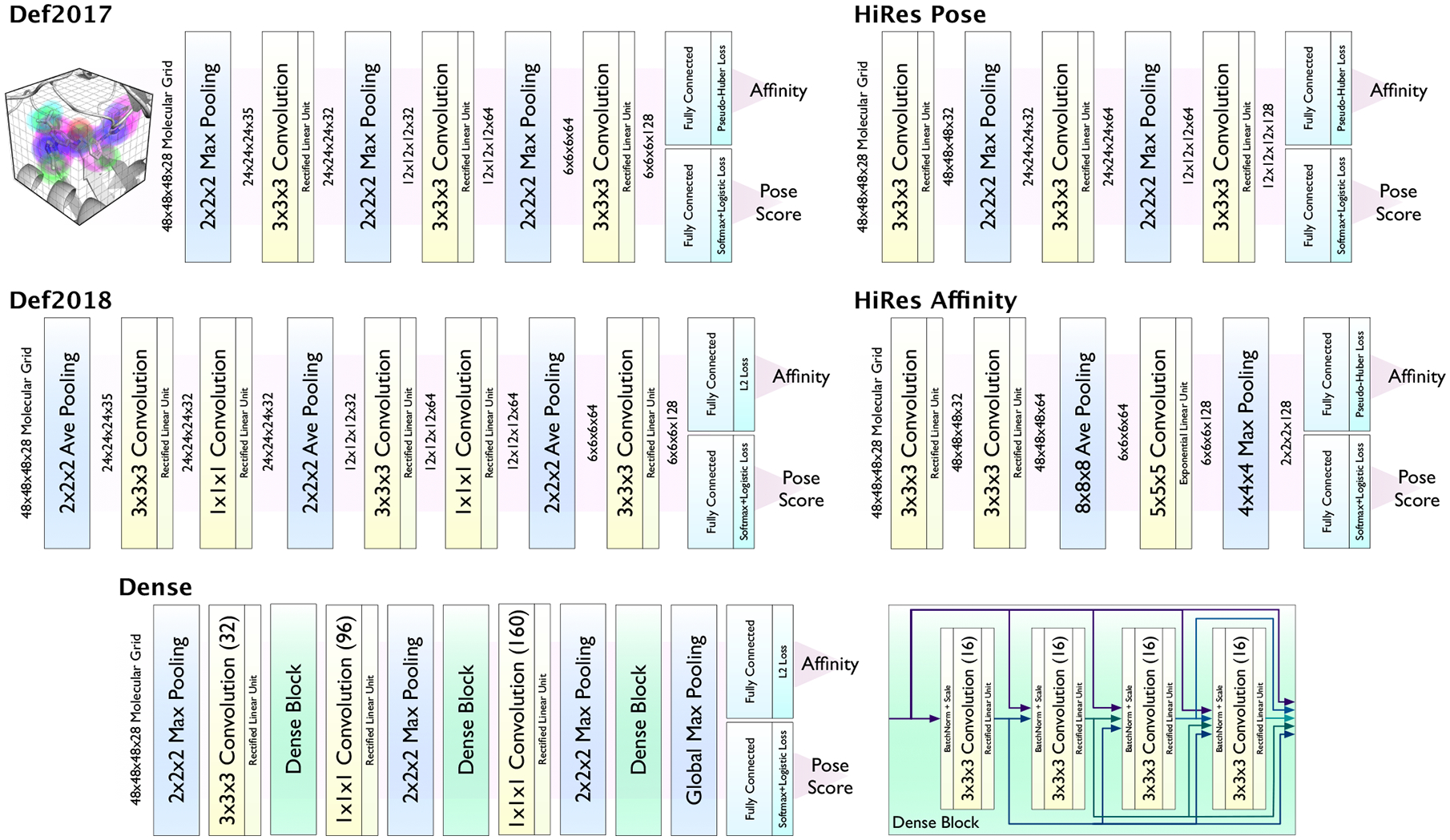
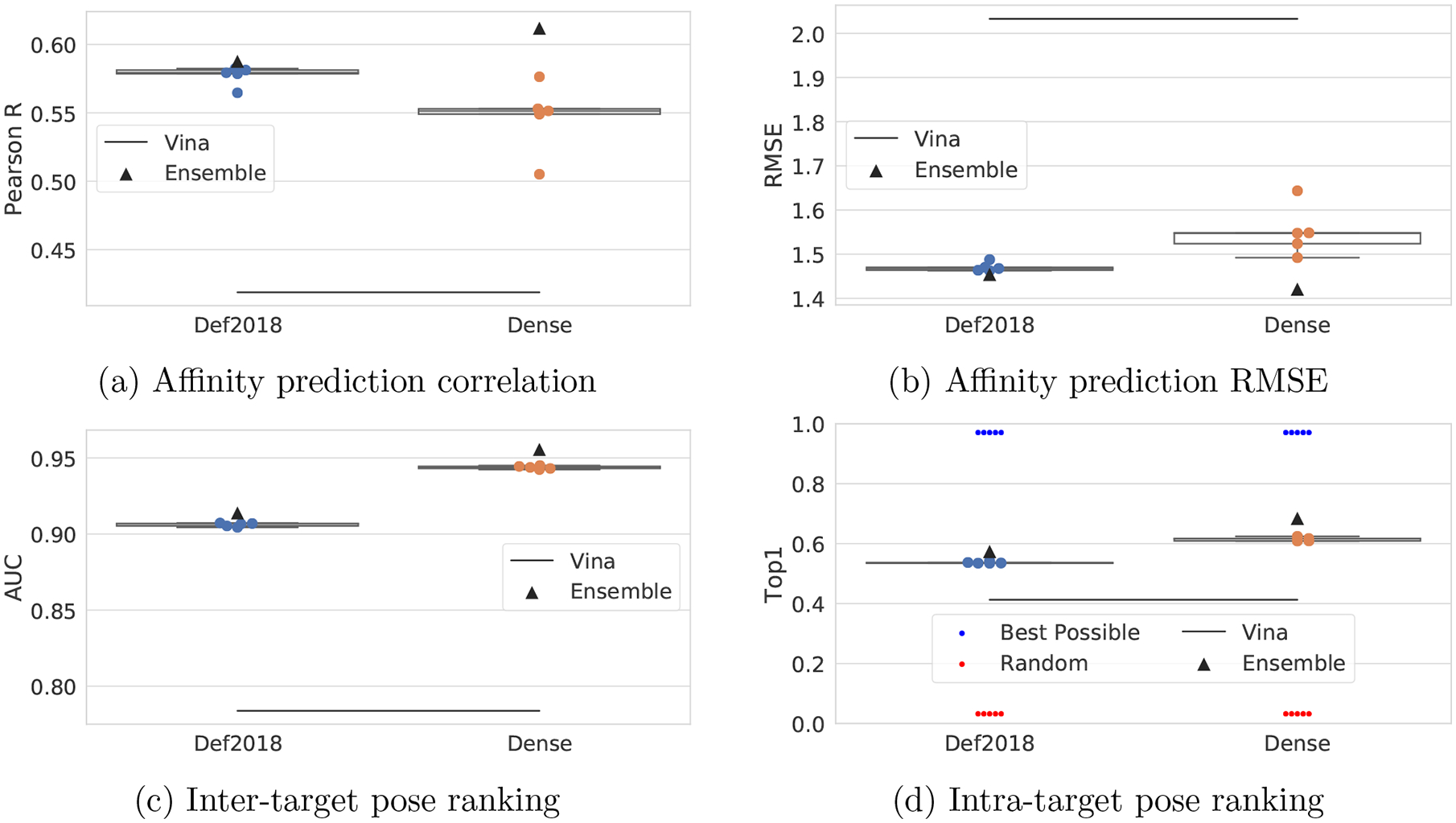
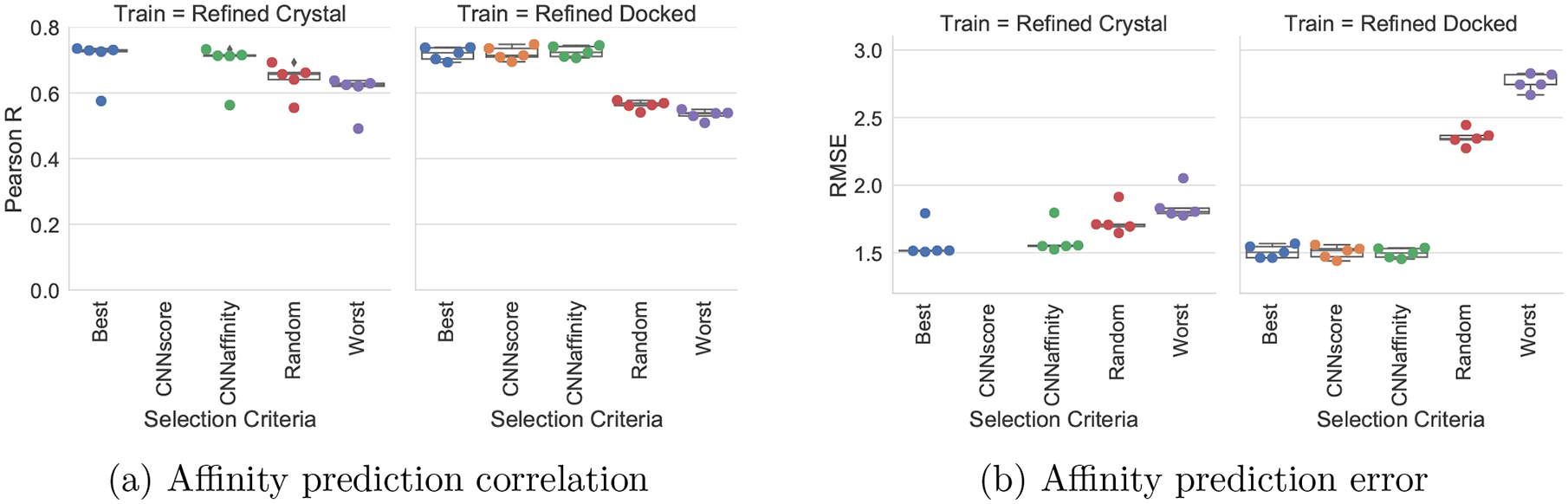
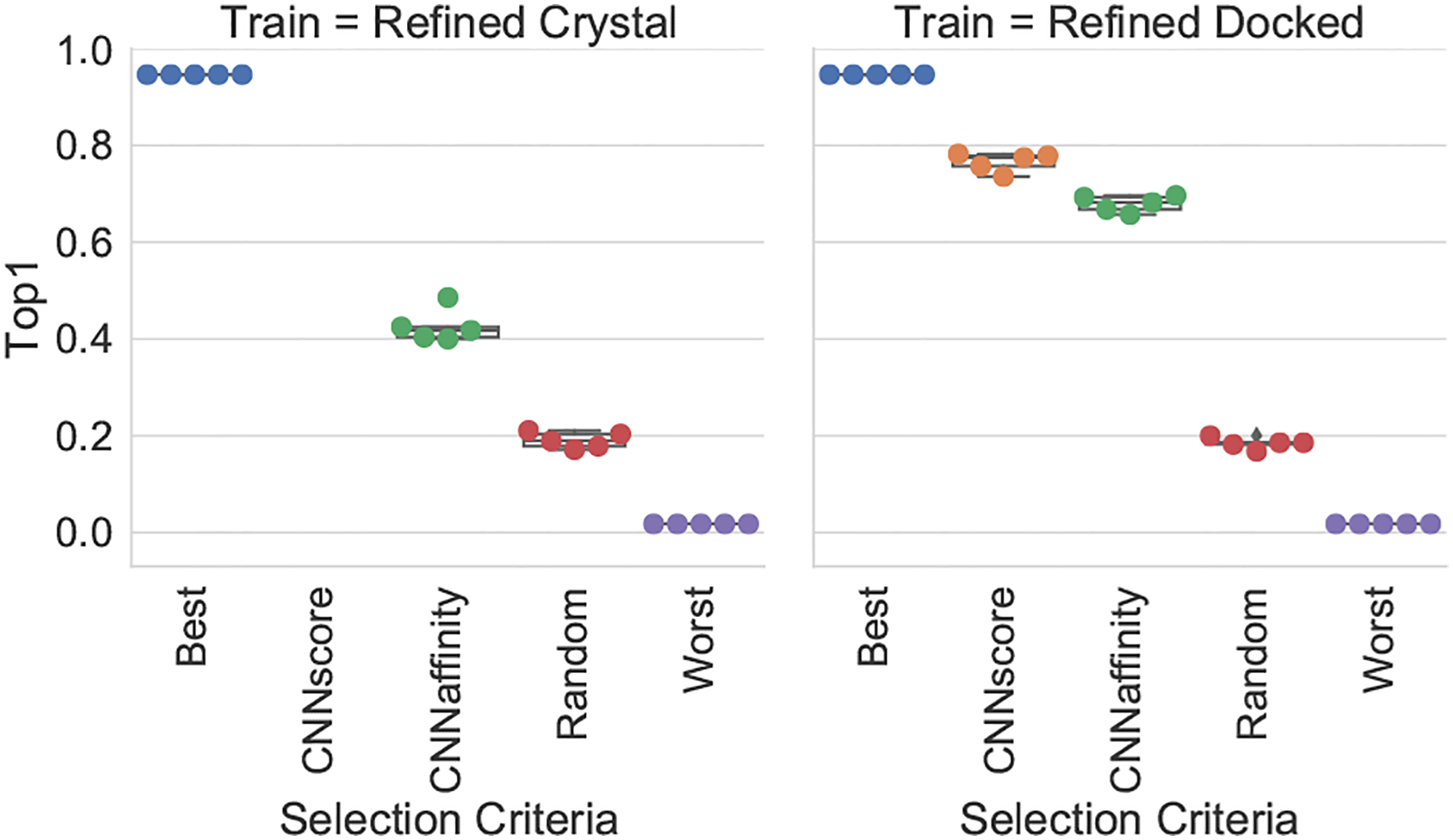
Francoeur Paul G, Masuda Tomohide, Sunseri Jocelyn, Jia Andrew, Iovanisci Richard B, Snyder Ian, Koes David R
Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, United States.
J Chem Inf Model. 2020 Sep 28;60(9):4200-4215. doi: 10.1021/acs.jcim.0c00411. Epub 2020 Sep 10.
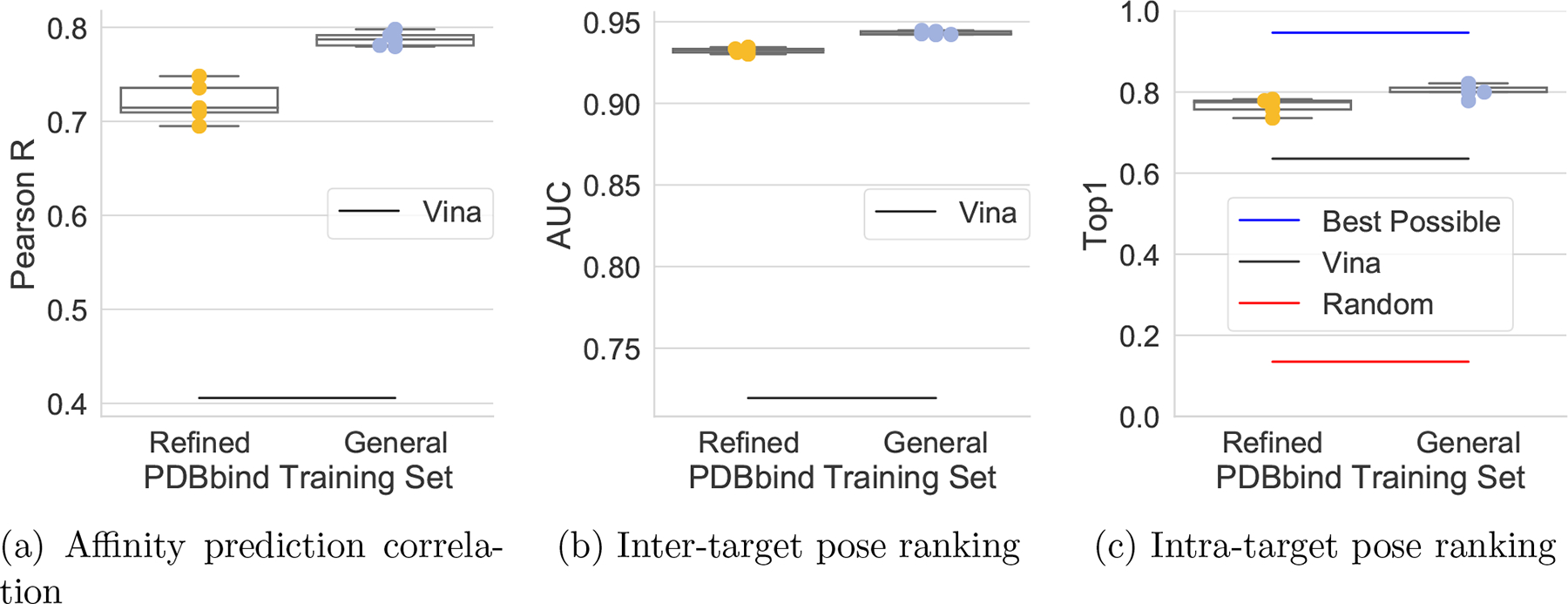
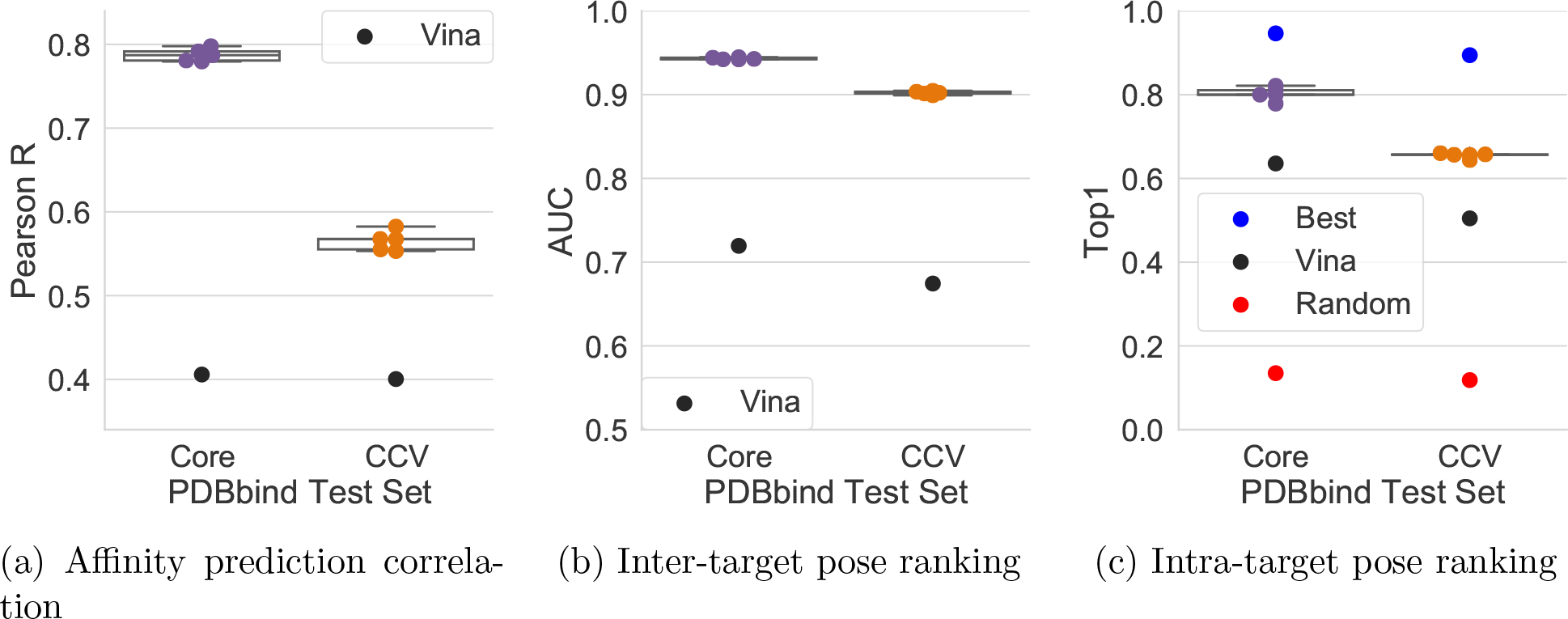
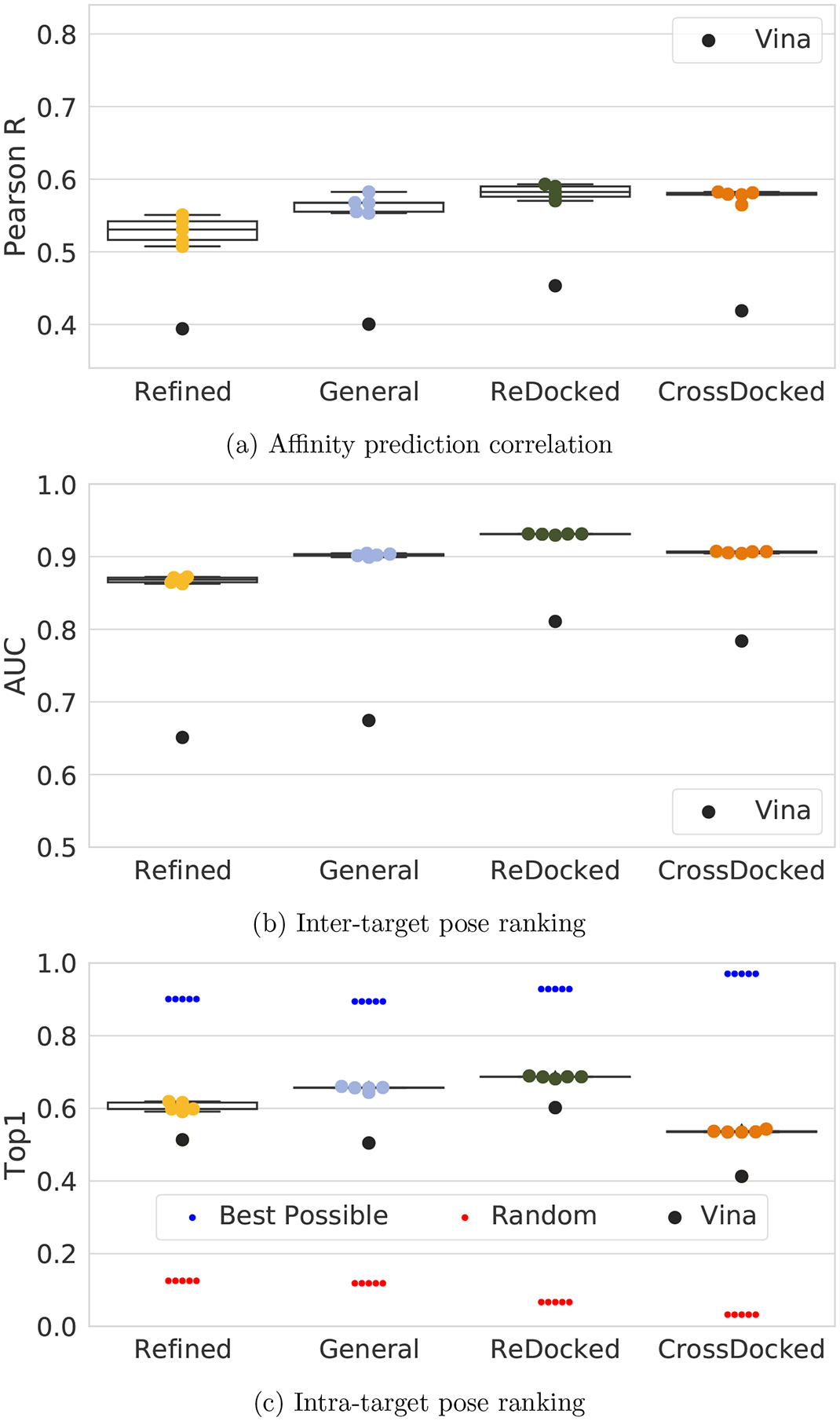
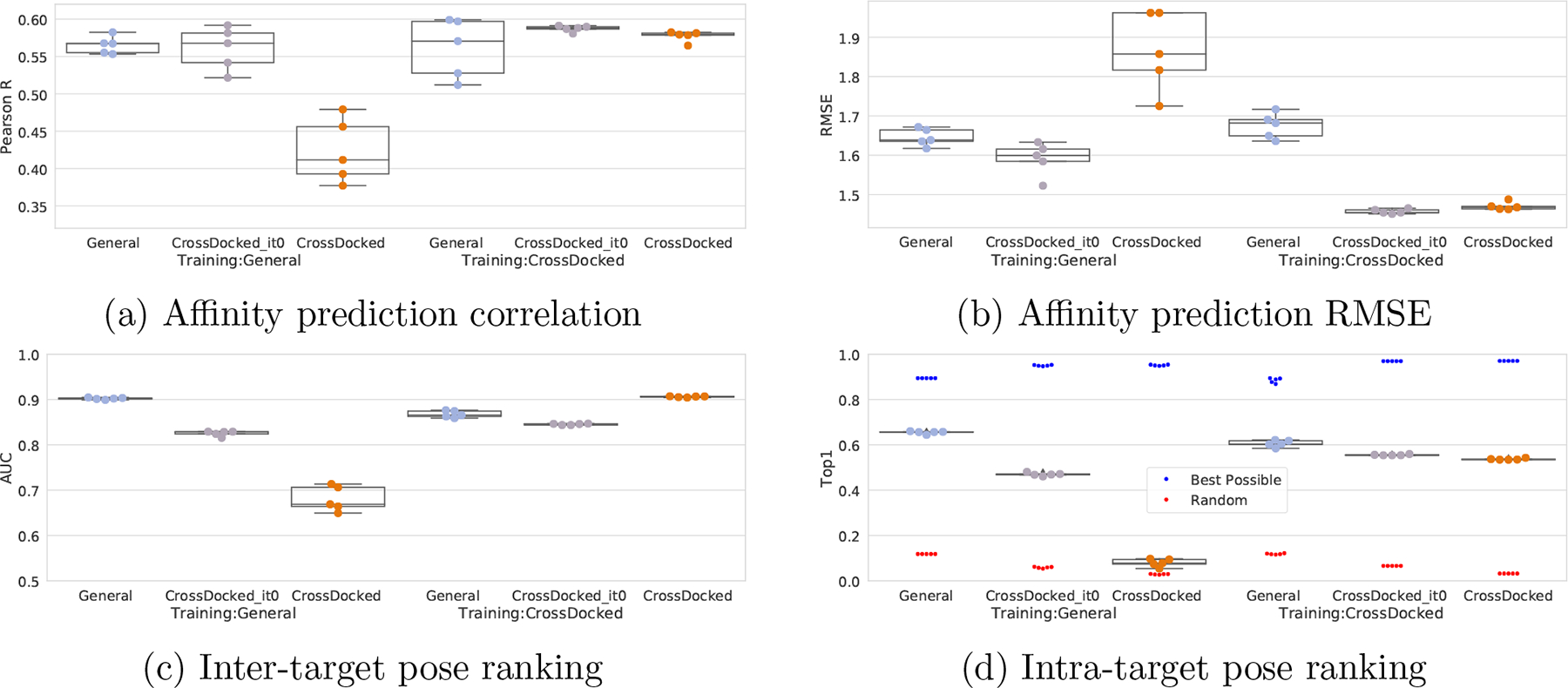
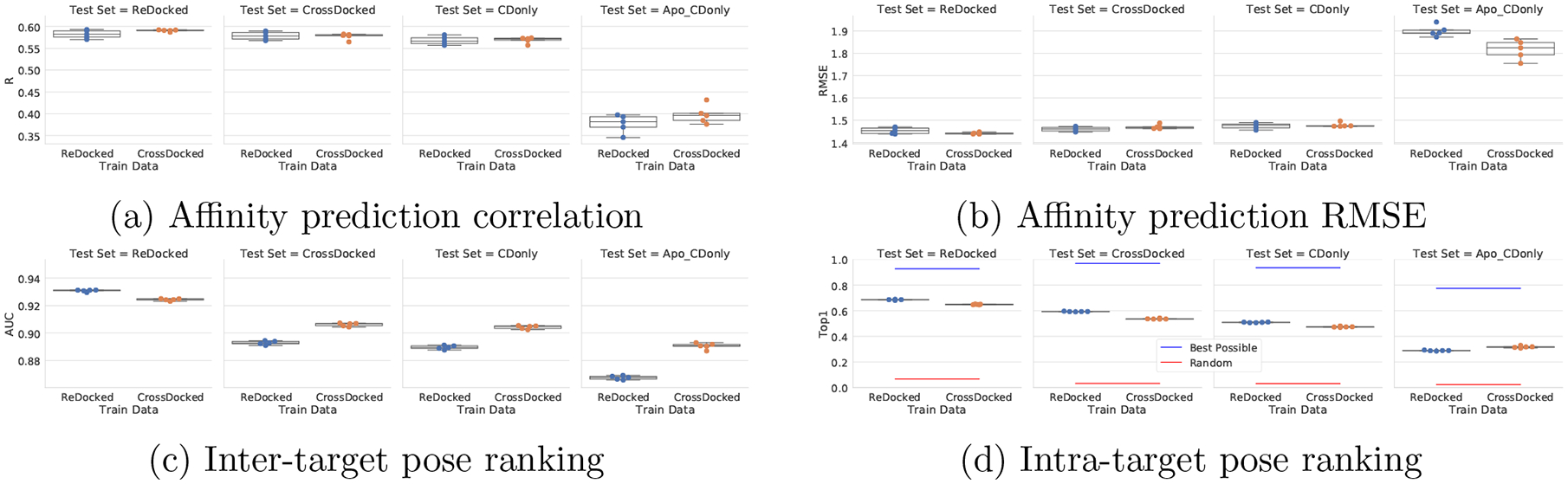
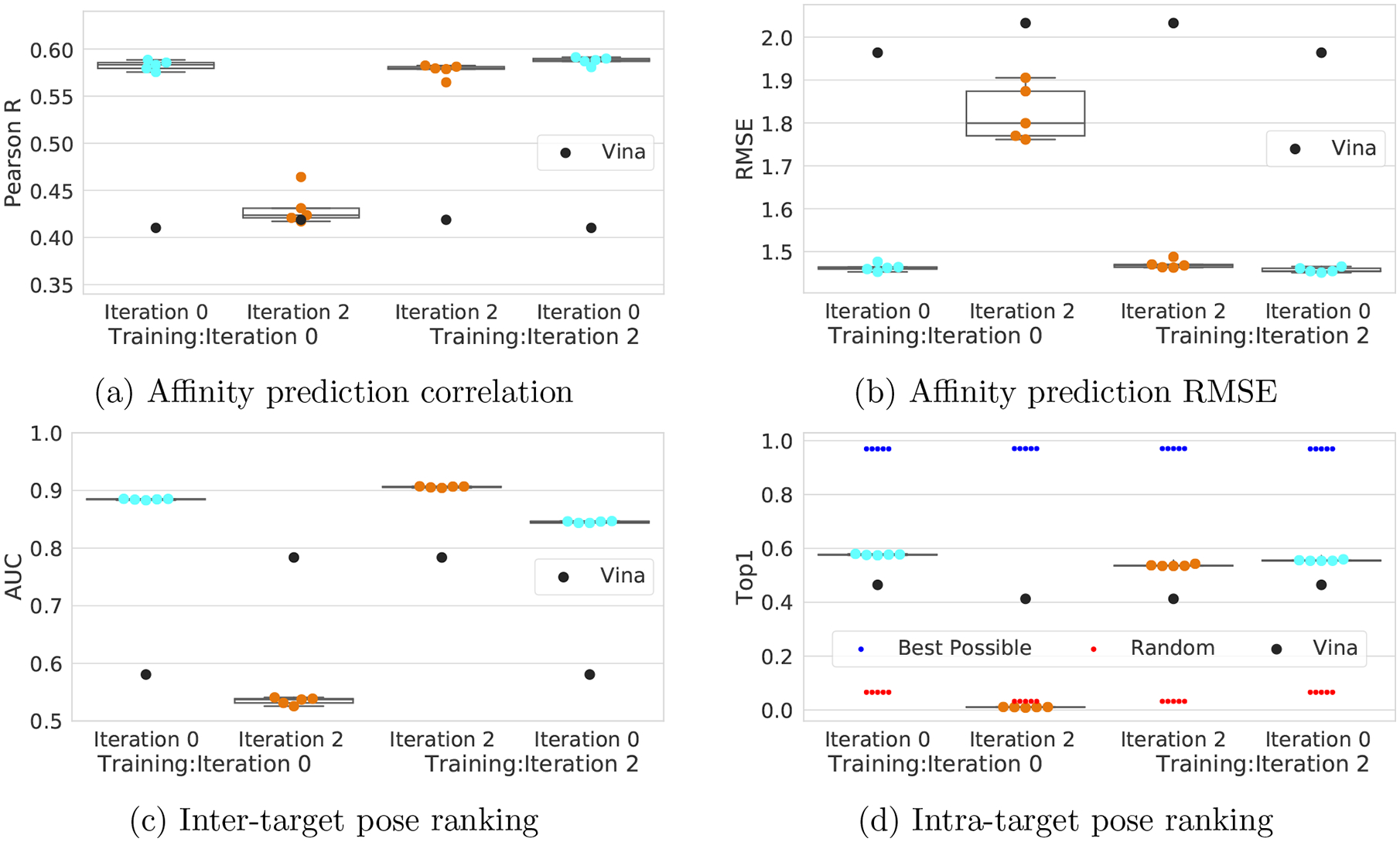
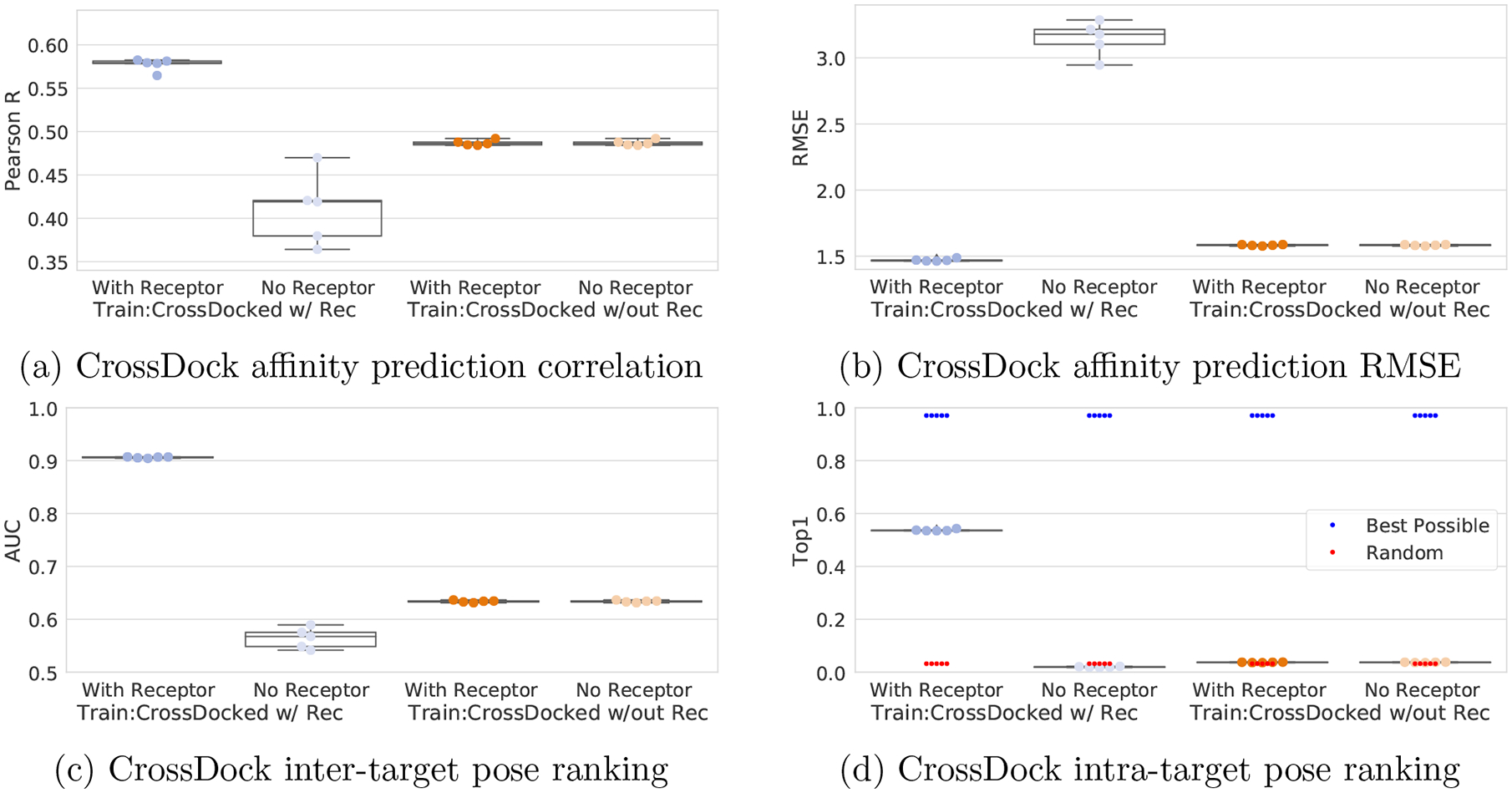
One of the main challenges in drug discovery is predicting protein-ligand binding affinity. Recently, machine learning approaches have made substantial progress on this task. However, current methods of model evaluation are overly optimistic in measuring generalization to new targets, and there does not exist a standard data set of sufficient size to compare performance between models. We present a new data set for structure-based machine learning, the CrossDocked2020 set, with 22.5 million poses of ligands docked into multiple similar binding pockets across the Protein Data Bank, and perform a comprehensive evaluation of grid-based convolutional neural network (CNN) models on this data set. We also demonstrate how the partitioning of the training data and test data can impact the results of models trained with the PDBbind data set, how performance improves by adding more lower-quality training data, and how training with docked poses imparts pose sensitivity to the predicted affinity of a complex. Our best performing model, an ensemble of five densely connected CNNs, achieves a root mean squared error of 1.42 and Pearson of 0.612 on the affinity prediction task, an AUC of 0.956 at binding pose classification, and a 68.4% accuracy at pose selection on the CrossDocked2020 set. By providing data splits for clustered cross-validation and the raw data for the CrossDocked2020 set, we establish the first standardized data set for training machine learning models to recognize ligands in noncognate target structures while also greatly expanding the number of poses available for training. In order to facilitate community adoption of this data set for benchmarking protein-ligand binding affinity prediction, we provide our models, weights, and the CrossDocked2020 set at https://github.com/gnina/models.
药物研发中的主要挑战之一是预测蛋白质-配体结合亲和力。最近,机器学习方法在这项任务上取得了重大进展。然而,当前的模型评估方法在衡量对新靶点的泛化能力时过于乐观,并且不存在足够大的标准数据集来比较模型之间的性能。我们提出了一个用于基于结构的机器学习的新数据集CrossDocked2020集,其中包含2250万个配体构象对接至蛋白质数据库中多个相似结合口袋的结果,并对基于网格的卷积神经网络(CNN)模型在该数据集上进行了全面评估。我们还展示了训练数据和测试数据的划分如何影响使用PDBbind数据集训练的模型的结果,增加更多质量较低的训练数据如何提高性能,以及使用对接构象进行训练如何使预测的复合物亲和力具有构象敏感性。我们表现最佳的模型是一个由五个密集连接的CNN组成的集成模型,在亲和力预测任务上实现了均方根误差为1.42,皮尔逊相关系数为0.612,在结合构象分类上的AUC为0.956,在CrossDocked2020集上的构象选择准确率为68.4%。通过提供用于聚类交叉验证的数据划分和CrossDocked2020集的原始数据,我们建立了第一个标准化数据集,用于训练机器学习模型以识别非同源靶标结构中的配体,同时也大大增加了可用于训练的构象数量。为了便于社区采用此数据集来基准测试蛋白质-配体结合亲和力预测,我们在https://github.com/gnina/models上提供了我们的模型、权重和CrossDocked2020集。