Reshef Yakir A, Rumker Laurie, Kang Joyce B, Nathan Aparna, Korsunsky Ilya, Asgari Samira, Murray Megan B, Moody D Branch, Raychaudhuri Soumya
Center for Data Sciences, Brigham and Women's Hospital, Boston, MA, USA.
Division of Genetics, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, USA.
Nat Biotechnol. 2022 Mar;40(3):355-363. doi: 10.1038/s41587-021-01066-4. Epub 2021 Oct 21.
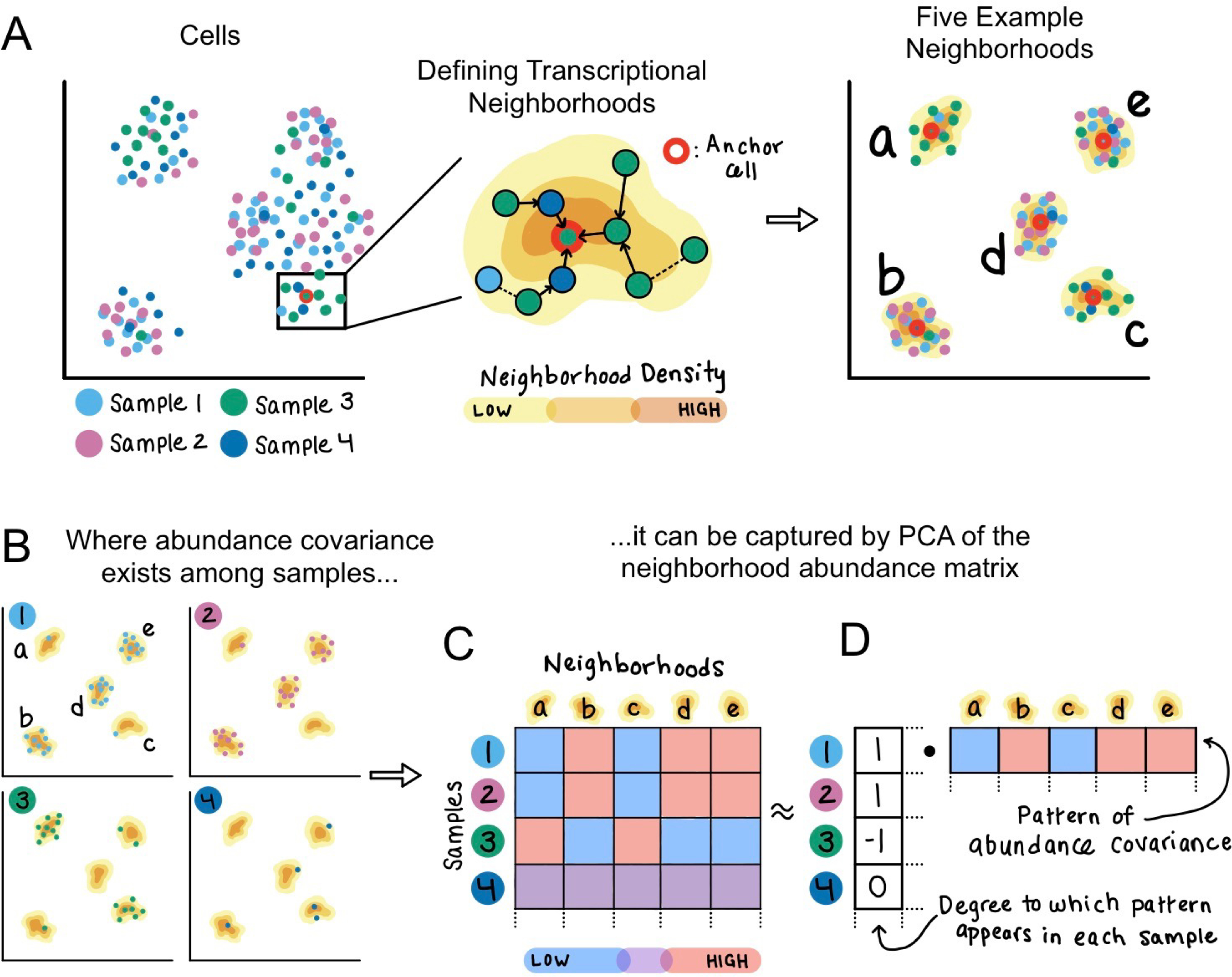
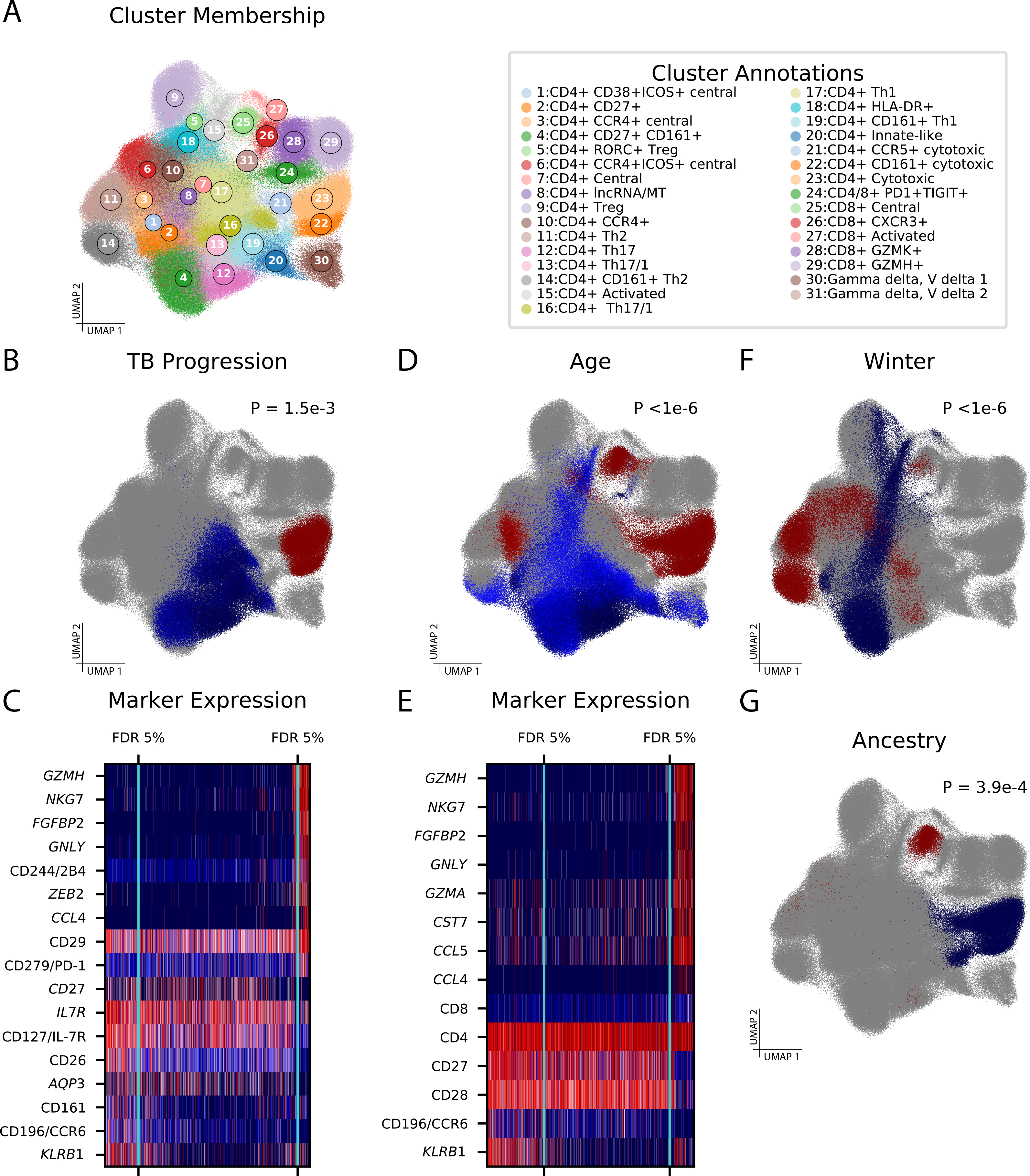
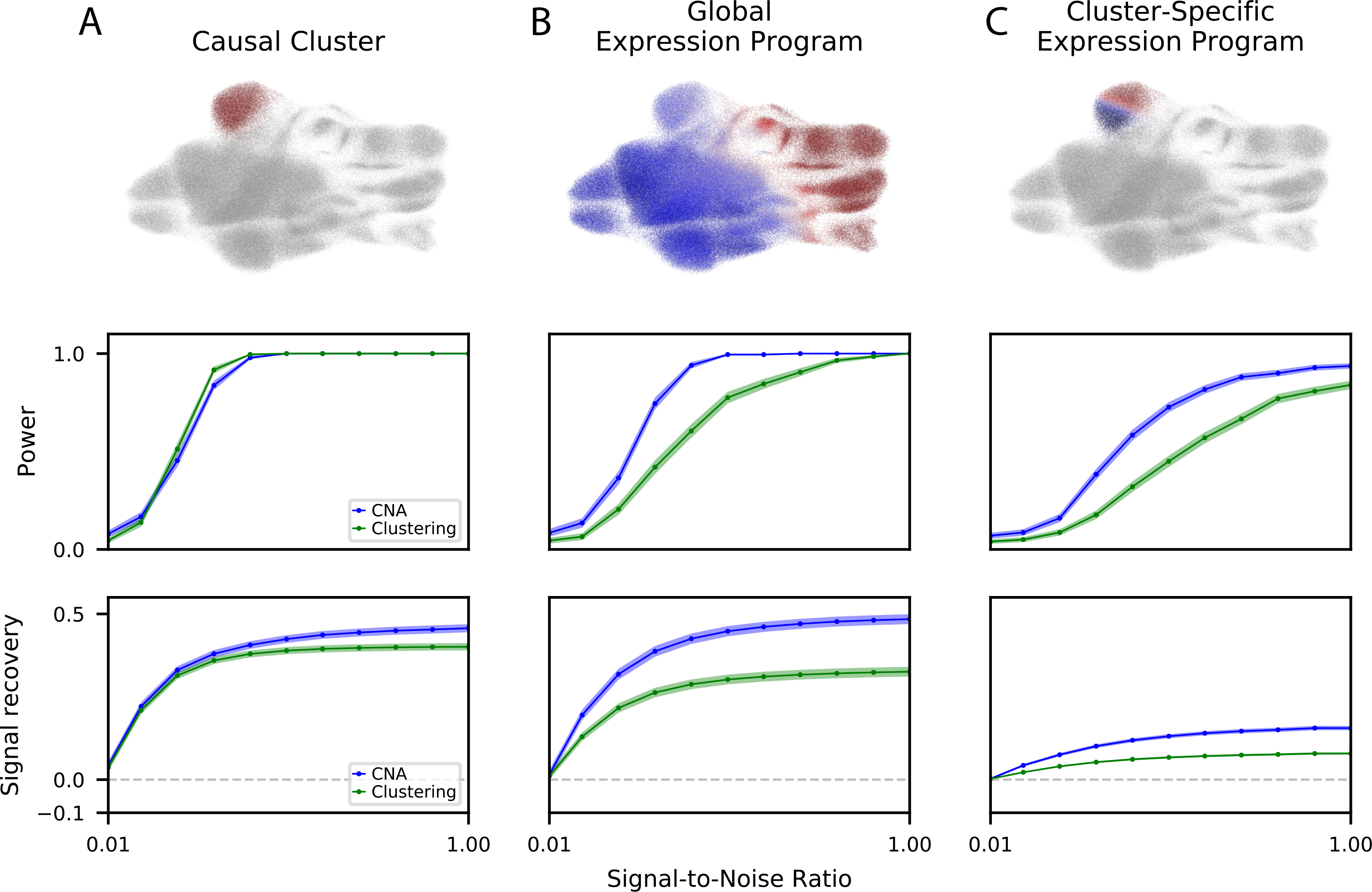
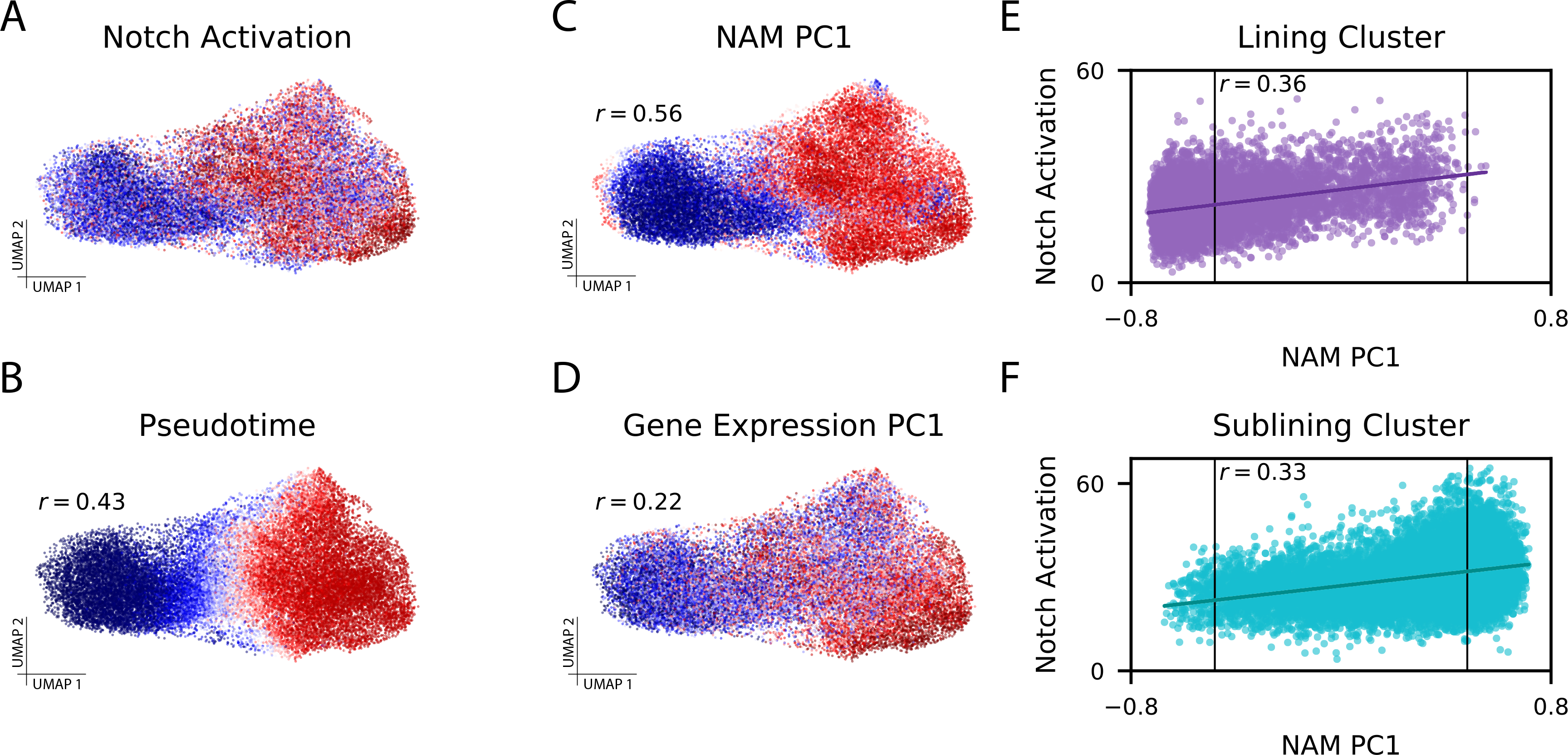
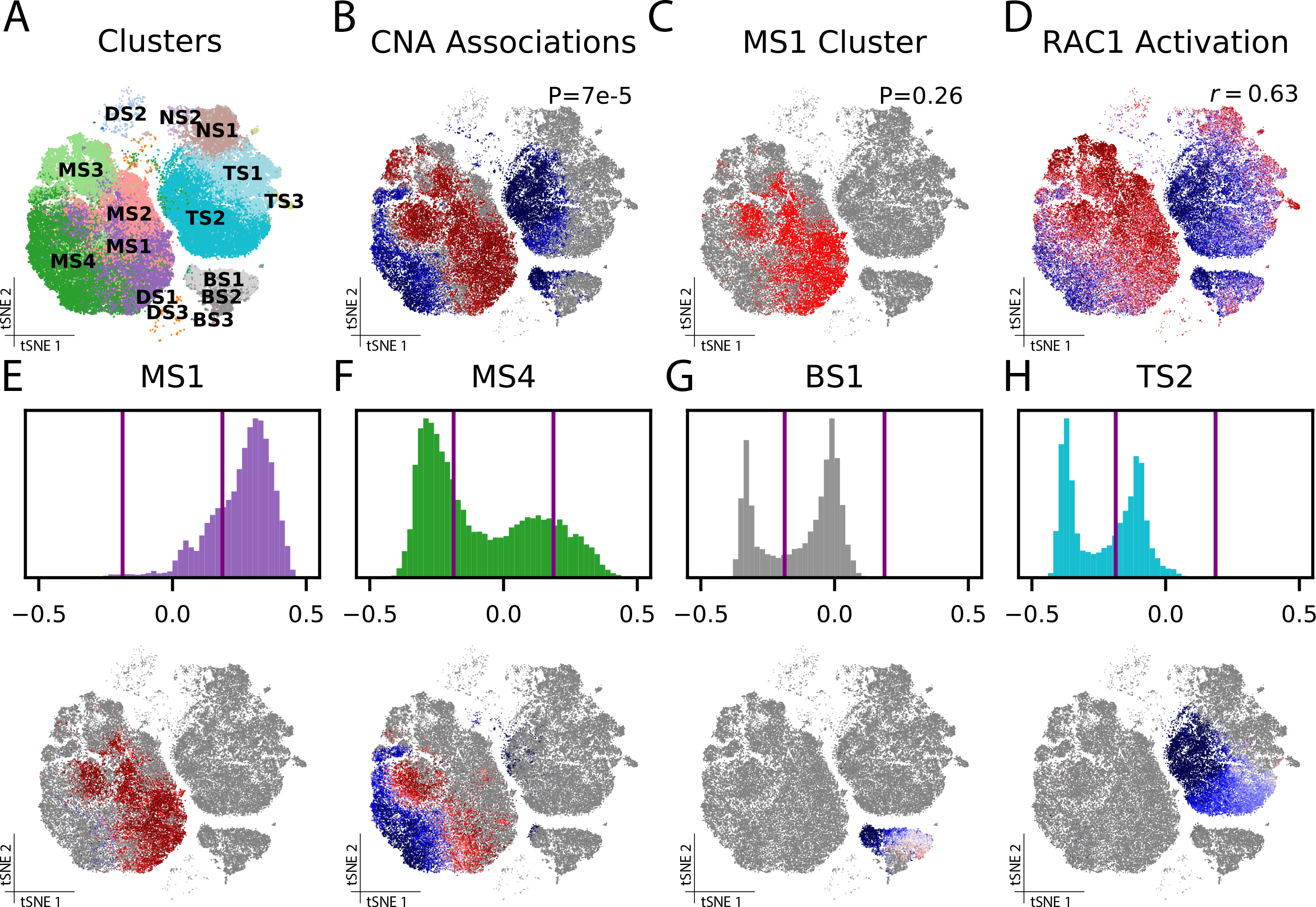
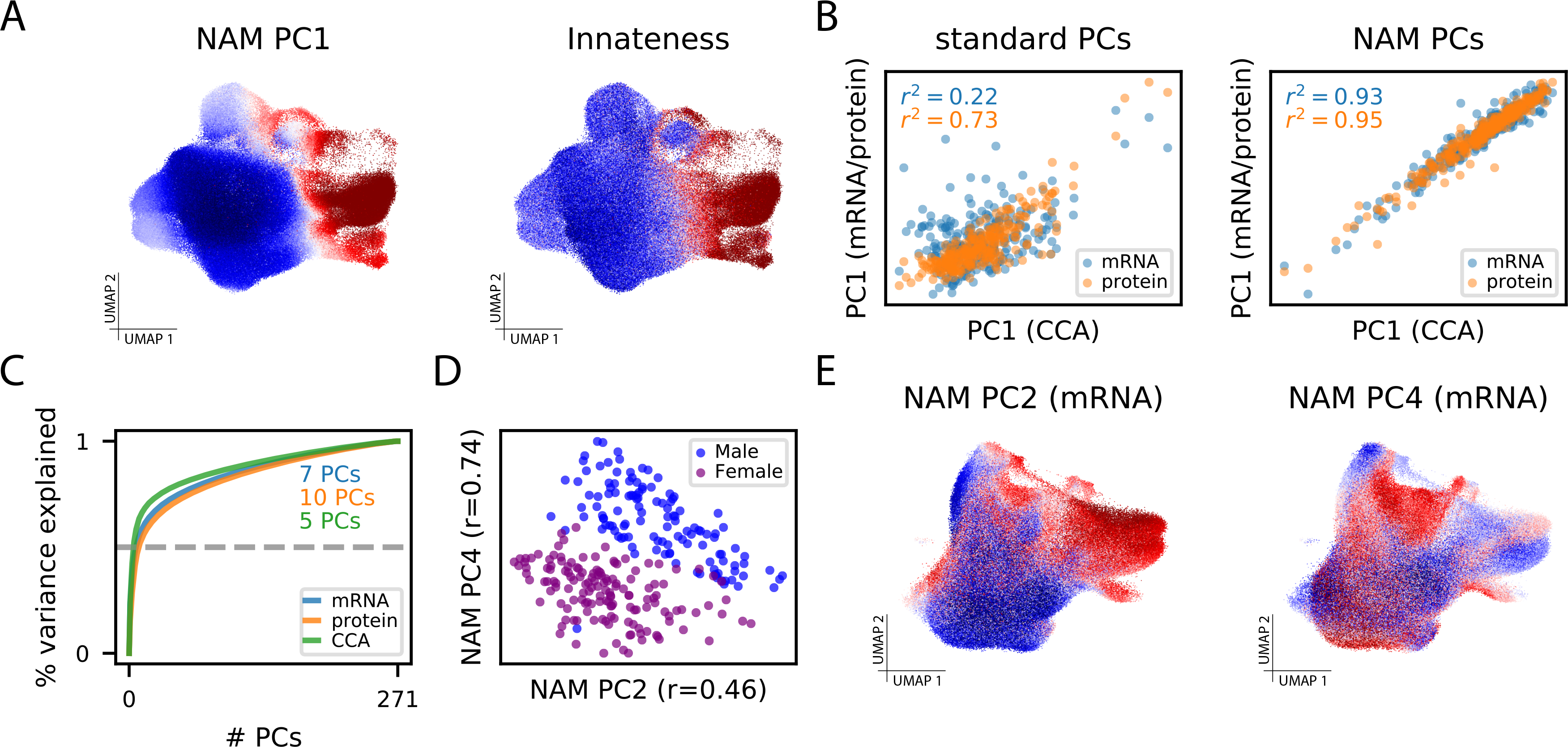
As single-cell datasets grow in sample size, there is a critical need to characterize cell states that vary across samples and associate with sample attributes, such as clinical phenotypes. Current statistical approaches typically map cells to clusters and then assess differences in cluster abundance. Here we present co-varying neighborhood analysis (CNA), an unbiased method to identify associated cell populations with greater flexibility than cluster-based approaches. CNA characterizes dominant axes of variation across samples by identifying groups of small regions in transcriptional space-termed neighborhoods-that co-vary in abundance across samples, suggesting shared function or regulation. CNA performs statistical testing for associations between any sample-level attribute and the abundances of these co-varying neighborhood groups. Simulations show that CNA enables more sensitive and accurate identification of disease-associated cell states than a cluster-based approach. When applied to published datasets, CNA captures a Notch activation signature in rheumatoid arthritis, identifies monocyte populations expanded in sepsis and identifies a novel T cell population associated with progression to active tuberculosis.
随着单细胞数据集样本量的增加,迫切需要对跨样本变化并与样本属性(如临床表型)相关的细胞状态进行表征。当前的统计方法通常将细胞映射到聚类中,然后评估聚类丰度的差异。在此,我们提出了共变邻域分析(CNA),这是一种无偏方法,用于识别相关细胞群体,比基于聚类的方法具有更大的灵活性。CNA通过识别转录空间中称为邻域的小区域组来表征跨样本的主要变异轴,这些邻域在样本间丰度共变,表明具有共同的功能或调控。CNA对任何样本水平属性与这些共变邻域组的丰度之间的关联进行统计检验。模拟表明,与基于聚类的方法相比,CNA能够更灵敏、准确地识别与疾病相关的细胞状态。当应用于已发表的数据集时,CNA在类风湿性关节炎中捕获了Notch激活特征,识别出脓毒症中扩增的单核细胞群体,并识别出与活动性肺结核进展相关的新型T细胞群体。