Prakash Amol, Taylor Lorne, Varkey Manu, Hoxie Nate, Mohammed Yassene, Goo Young Ah, Peterman Scott, Moghekar Abhay, Yuan Yuting, Glaros Trevor, Steele Joel R, Faridi Pouya, Parihari Shashwati, Srivastava Sanjeeva, Otto Joseph J, Nyalwidhe Julius O, Semmes O John, Moran Michael F, Madugundu Anil, Mun Dong Gi, Pandey Akhilesh, Mahoney Keira E, Shabanowitz Jeffrey, Saxena Satya, Orsburn Benjamin C
Optys Tech Corporation, Shrewsbury, MA 01545, USA.
McGill University Health Center, Montreal, QC H4A 3J1, Canada.
Cancers (Basel). 2021 Oct 9;13(20):5034. doi: 10.3390/cancers13205034.
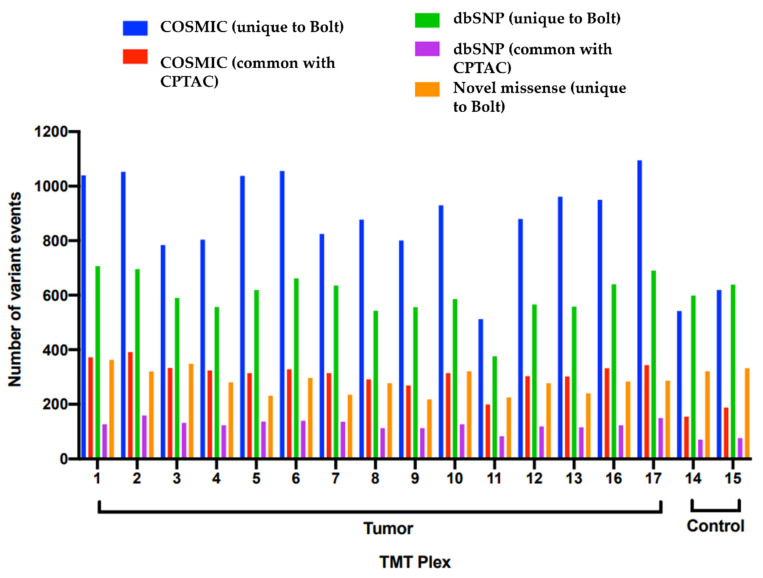
The Clinical Proteomic Tumor Analysis Consortium (CPTAC) has provided some of the most in-depth analyses of the phenotypes of human tumors ever constructed. Today, the majority of proteomic data analysis is still performed using software housed on desktop computers which limits the number of sequence variants and post-translational modifications that can be considered. The original CPTAC studies limited the search for PTMs to only samples that were chemically enriched for those modified peptides. Similarly, the only sequence variants considered were those with strong evidence at the exon or transcript level. In this multi-institutional collaborative reanalysis, we utilized unbiased protein databases containing millions of human sequence variants in conjunction with hundreds of common post-translational modifications. Using these tools, we identified tens of thousands of high-confidence PTMs and sequence variants. We identified 4132 phosphorylated peptides in nonenriched samples, 93% of which were confirmed in the samples which were chemically enriched for phosphopeptides. In addition, our results also cover 90% of the high-confidence variants reported by the original proteogenomics study, without the need for sample specific next-generation sequencing. Finally, we report fivefold more somatic and germline variants that have an independent evidence at the peptide level, including mutations in ERRB2 and BCAS1. In this reanalysis of CPTAC proteomic data with cloud computing, we present an openly available and searchable web resource of the highest-coverage proteomic profiling of human tumors described to date.
临床蛋白质组肿瘤分析联盟(CPTAC)对人类肿瘤表型进行了一些有史以来最深入的分析。如今,大多数蛋白质组数据分析仍使用台式计算机上的软件进行,这限制了可考虑的序列变异和翻译后修饰的数量。CPTAC的原始研究将翻译后修饰的搜索仅限于那些化学富集修饰肽的样本。同样,唯一考虑的序列变异是那些在外显子或转录水平有确凿证据的变异。在这项多机构合作的重新分析中,我们利用了包含数百万个人类序列变异以及数百种常见翻译后修饰的无偏蛋白质数据库。使用这些工具,我们鉴定出了数以万计的高可信度翻译后修饰和序列变异。我们在未富集的样本中鉴定出4132个磷酸化肽,其中93%在化学富集磷酸肽的样本中得到证实。此外,我们的结果还涵盖了原始蛋白质基因组学研究报告的90%的高可信度变异,而无需进行样本特异性的下一代测序。最后,我们报告了在肽水平有独立证据的体细胞和种系变异数量增加了五倍,包括ERBB2和BCAS1中的突变。在这项利用云计算对CPTAC蛋白质组数据进行的重新分析中,我们展示了一个公开可用且可搜索的网络资源,它是迄今为止所描述的人类肿瘤最高覆盖率蛋白质组分析。