Marques Andrew D, Sherrill-Mix Scott, Everett John, Reddy Shantan, Hokama Pascha, Roche Aoife M, Hwang Young, Glascock Abigail, Whiteside Samantha A, Graham-Wooten Jevon, Khatib Layla A, Fitzgerald Ayannah S, Moustafa Ahmed M, Bianco Colleen, Rajagopal Swetha, Helton Jenna, Deming Regan, Denu Lidiya, Ahmed Azad, Kitt Eimear, Coffin Susan E, Newbern Claire, Mell Josh Chang, Planet Paul J, Badjatia Nitika, Richards Bonnie, Wang Zi-Xuan, Cannuscio Carolyn C, Strelau Katherine M, Jaskowiak-Barr Anne, Cressman Leigh, Loughrey Sean, Ganguly Arupa, Feldman Michael D, Collman Ronald G, Rodino Kyle G, Kelly Brendan J, Bushman Frederic D
Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA.
Pulmonary, Allergy and Critical Care Division; Department of Medicine; University of Pennsylvania Perelman School of Medicine; Philadelphia, PA.
medRxiv. 2021 Nov 17:2021.10.18.21264623. doi: 10.1101/2021.10.18.21264623.
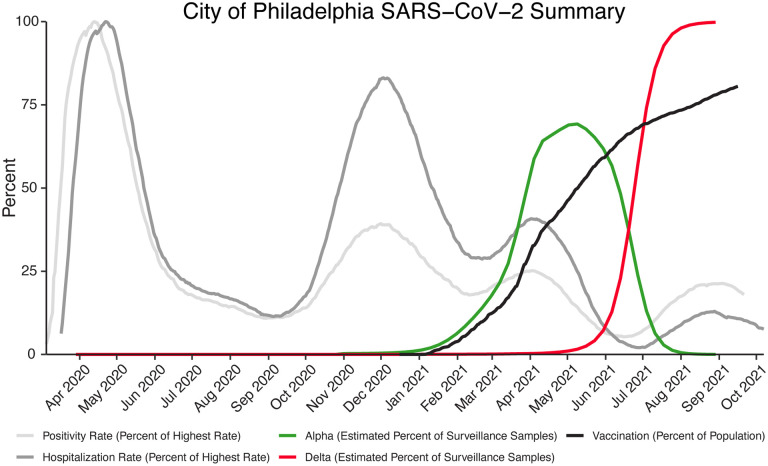
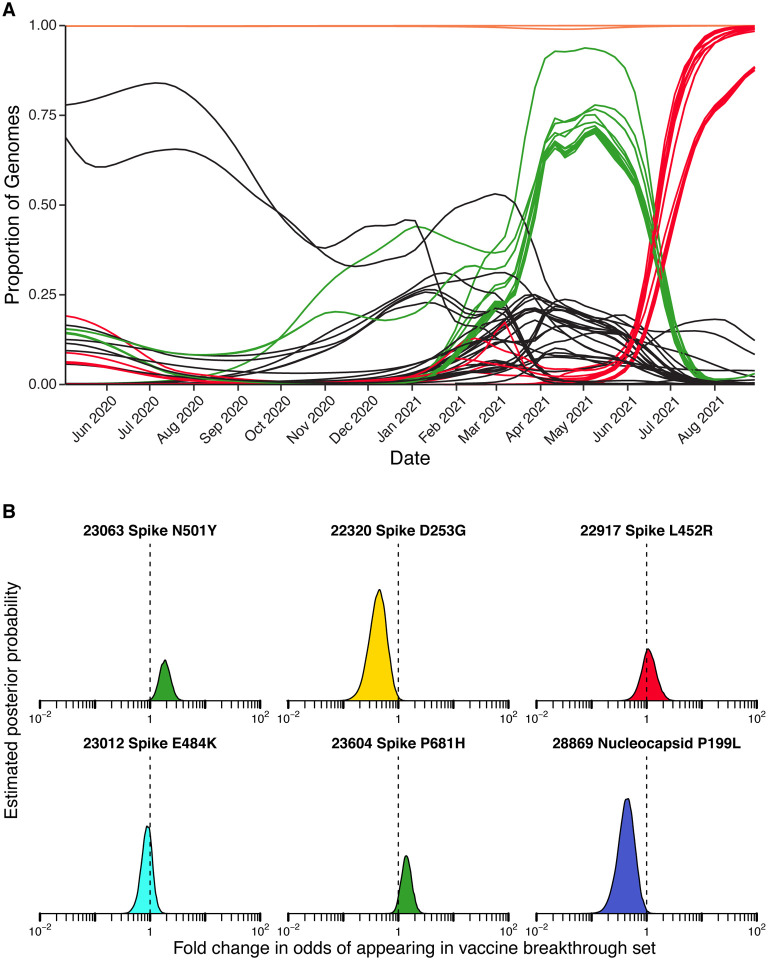
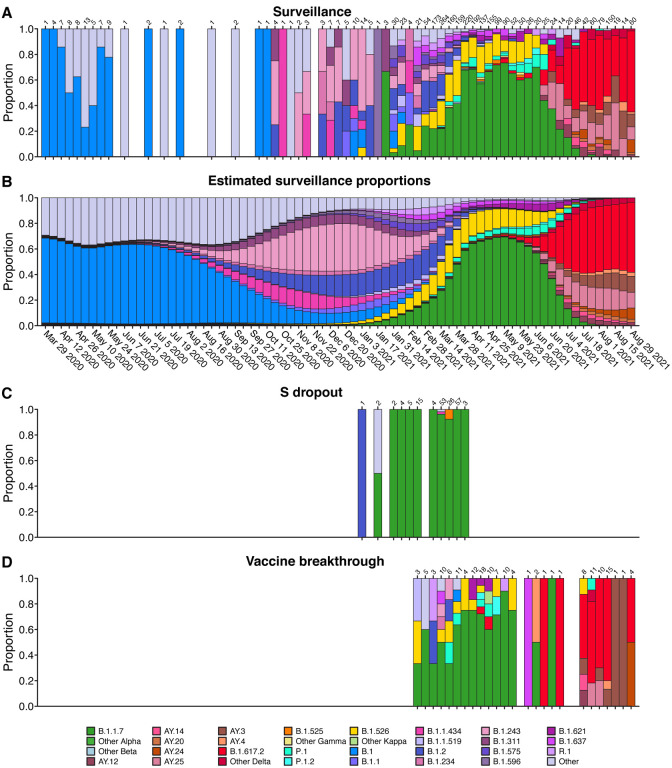
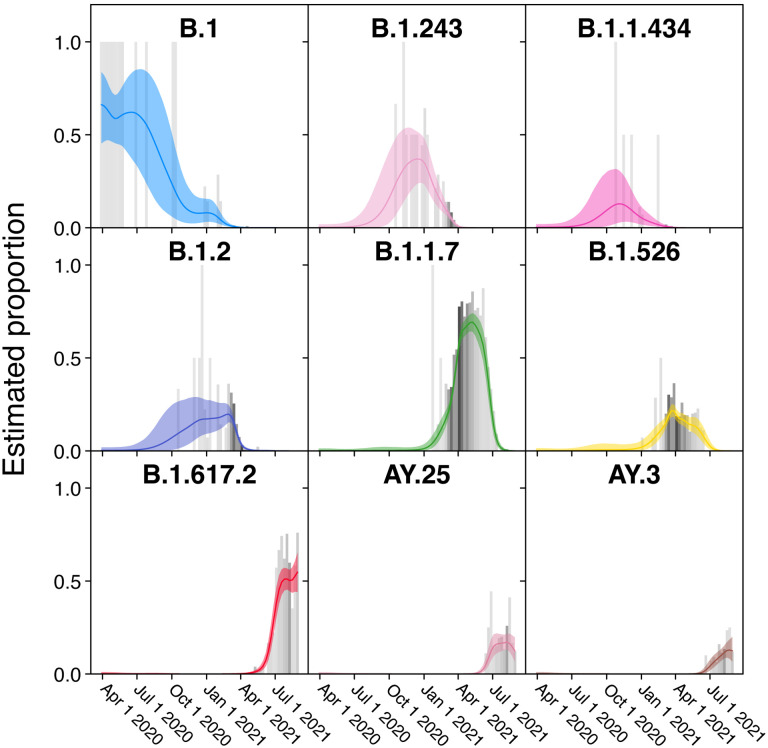
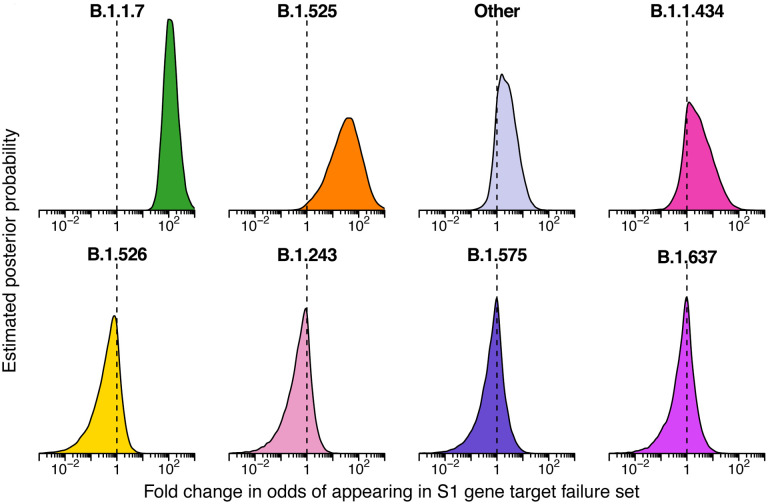
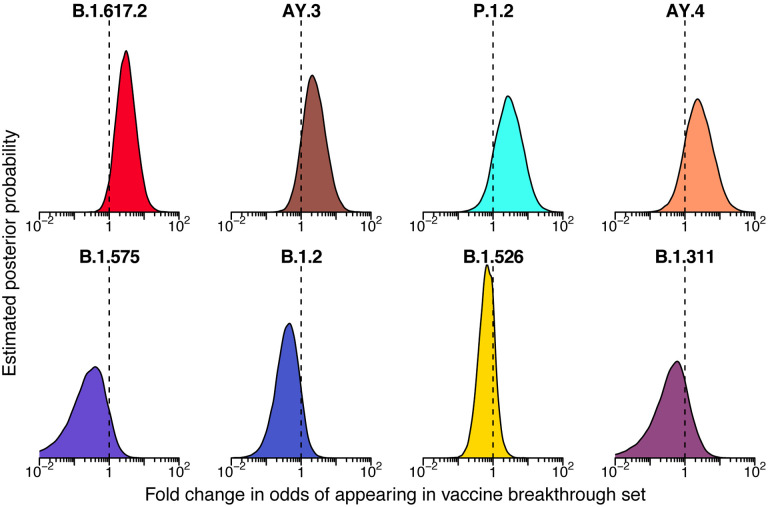
The severe acute respiratory coronavirus-2 (SARS-CoV-2) is the cause of the global outbreak of COVID-19. Evidence suggests that the virus is evolving to allow efficient spread through the human population, including vaccinated individuals. Here we report a study of viral variants from surveillance of the Delaware Valley, including the city of Philadelphia, and variants infecting vaccinated subjects. We sequenced and analyzed complete viral genomes from 2621 surveillance samples from March 2020 to September 2021 and compared them to genome sequences from 159 vaccine breakthroughs. In the early spring of 2020, all detected variants were of the B.1 and closely related lineages. A mixture of lineages followed, notably including B.1.243 followed by B.1.1.7 (alpha), with other lineages present at lower levels. Later isolations were dominated by B.1.617.2 (delta) and other delta lineages; delta was the exclusive variant present by the last time sampled. To investigate whether any variants appeared preferentially in vaccine breakthroughs, we devised a model based on Bayesian autoregressive moving average logistic multinomial regression to allow rigorous comparison. This revealed that B.1.617.2 (delta) showed three-fold enrichment in vaccine breakthrough cases (odds ratio of 3; 95% credible interval 0.89-11). Viral point substitutions could also be associated with vaccine breakthroughs, notably the N501Y substitution found in the alpha, beta and gamma variants (odds ratio 2.04; 95% credible interval of 1.25-3.18). This study thus provides a detailed picture of viral evolution in the Delaware Valley and a geographically matched analysis of vaccine breakthroughs; it also introduces a rigorous statistical approach to interrogating enrichment of viral variants.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)是导致全球新冠疫情大流行的病原体。有证据表明,该病毒正在不断进化,以便在包括接种疫苗的人群在内的人类群体中高效传播。在此,我们报告了一项对特拉华谷(包括费城)监测到的病毒变体以及感染接种疫苗者的变体的研究。我们对2020年3月至2021年9月期间2621份监测样本中的完整病毒基因组进行了测序和分析,并将其与159例疫苗突破感染病例的基因组序列进行了比较。2020年早春,所有检测到的变体均属于B.1及其密切相关的谱系。随后出现了多种谱系的混合,其中特别值得注意的是B.1.243,随后是B.1.1.7(阿尔法),其他谱系的占比相对较低。后来的分离株主要是B.1.617.2(德尔塔)和其他德尔塔谱系;在最后一次采样时,德尔塔是唯一存在的变体。为了研究是否有任何变体在疫苗突破感染病例中出现的比例偏高,我们设计了一个基于贝叶斯自回归移动平均逻辑多项回归的模型,以便进行严格比较。结果显示,B.1.617.2(德尔塔)在疫苗突破感染病例中的富集程度为三倍(优势比为3;95%可信区间为0.89 - 11)。病毒的点突变也可能与疫苗突破感染有关,特别是在阿尔法、贝塔和伽马变体中发现的N501Y突变(优势比为2.04;95%可信区间为1.25 - 3.18)。因此,本研究详细描绘了特拉华谷的病毒进化情况,并对疫苗突破感染进行了地域匹配分析;同时还引入了一种严格的统计方法来探究病毒变体的富集情况。