Institute of Bacterial Infections and Zoonoses, Friedrich-Loeffler-Institut, Jena, Germany.
Department of Pathology, Faculty of Veterinary Medicine, Zagazig University, Zagazig, Sharkia Province, Egypt.
Microbiol Spectr. 2021 Oct 31;9(2):e0053321. doi: 10.1128/Spectrum.00533-21. Epub 2021 Oct 27.
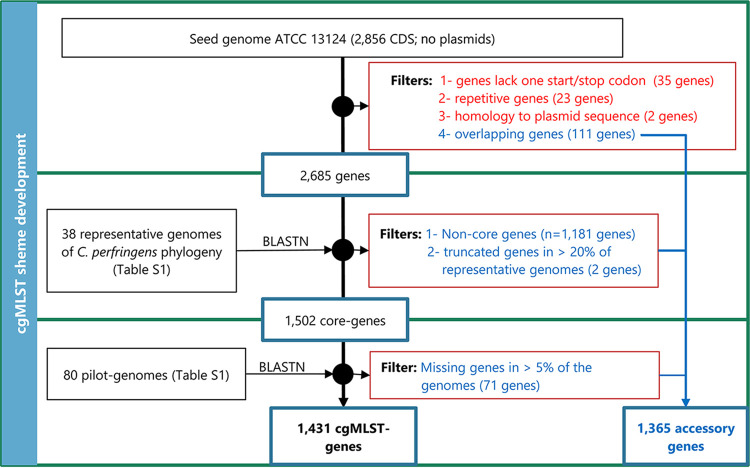
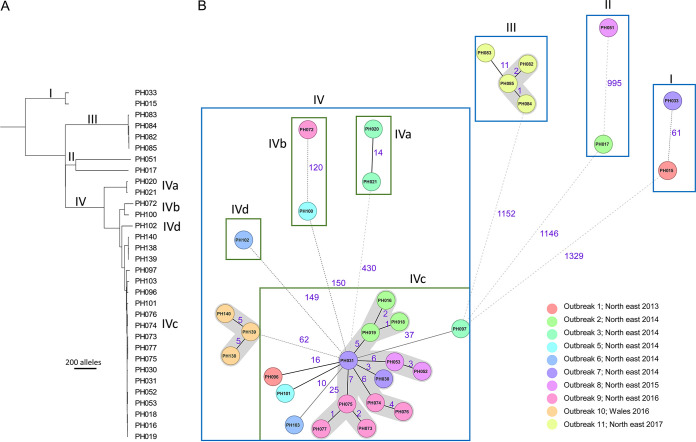
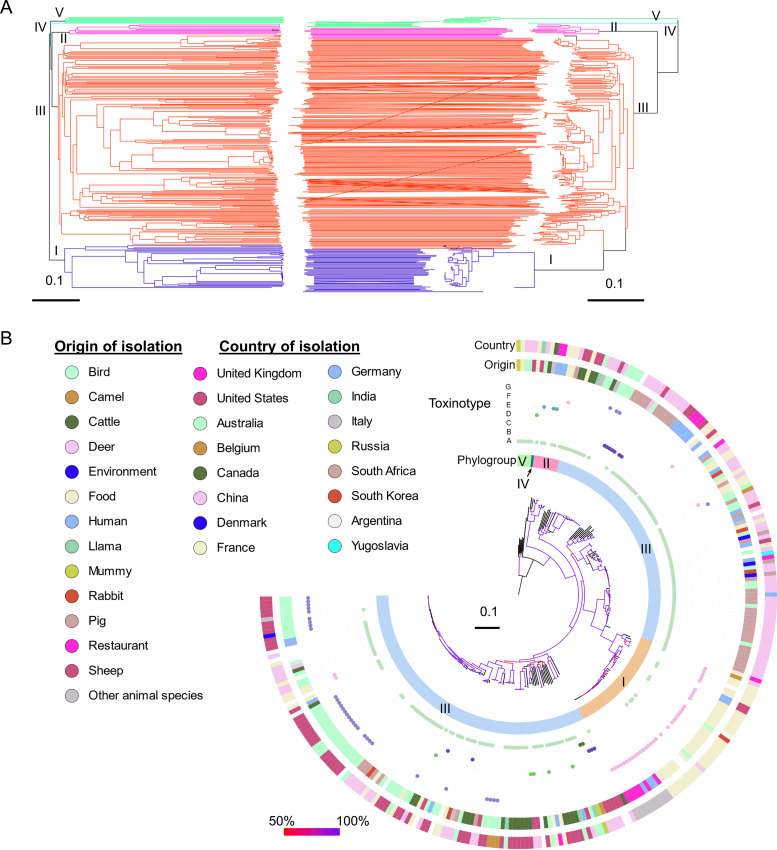
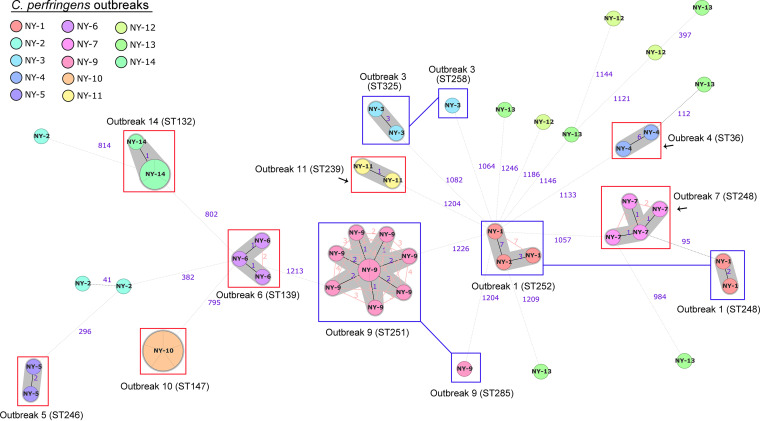
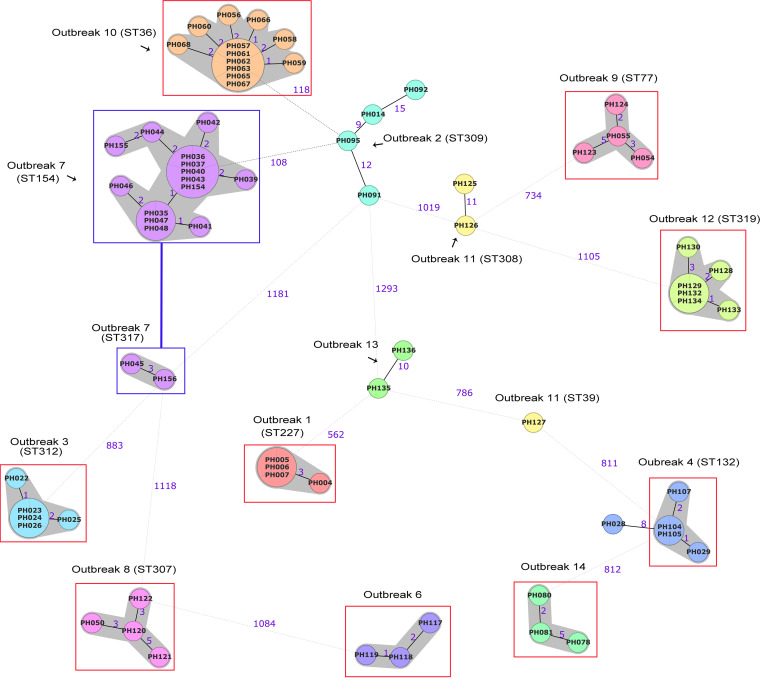
Clostridium perfringens is a spore-forming anaerobic pathogen responsible for a variety of histotoxic and intestinal infections in humans and animals. High-resolution genotyping aiming to identify bacteria at strain level has become increasingly important in modern microbiology to understand pathogen transmission pathways and to tackle infection sources. This study aimed at establishing a publicly available genome-wide multilocus sequence-typing (MLST) scheme for C. perfringens. A total of 1,431 highly conserved core genes (1.34 megabases; 50% of the reference genome genes) were indexed for a core genome-based MLST (cgMLST) scheme for C. perfringens. The scheme was applied to 282 ecologically and geographically diverse genomes, showing that the genotyping results of cgMLST were highly congruent with the core genome-based single-nucleotide-polymorphism typing in terms of resolution and tree topology. In addition, the cgMLST provided a greater discrimination than classical MLST methods for C. perfringens. The usability of the scheme for outbreak analysis was confirmed by reinvestigating published outbreaks of C. perfringens-associated infections in the United States and the United Kingdom. In summary, a publicly available scheme and an allele nomenclature database for genomic typing of C. perfringens have been established and can be used for broad-based and standardized epidemiological studies. Global epidemiological surveillance of bacterial pathogens is enhanced by the availability of standard tools and sharing of typing data. The use of whole-genome sequencing has opened the possibility for high-resolution characterization of bacterial strains down to the clonal and subclonal levels. Core genome multilocus sequence typing is a robust system that uses highly conserved core genes for deep genotyping. The method has been successfully and widely used to describe the epidemiology of various bacterial species. Nevertheless, a cgMLST typing scheme for Clostridium perfringens is currently not publicly available. In this study, we (i) developed a cgMLST typing scheme for C. perfringens, (ii) evaluated the performance of the scheme on different sets of C. perfringens genomes from different hosts and geographic regions as well as from different outbreak situations, and, finally, (iii) made this scheme publicly available supported by an allele nomenclature database for global and standard genomic typing.
产气荚膜梭菌是一种产芽孢的厌氧病原体,可导致人类和动物的多种组织毒性和肠道感染。高分辨率基因分型旨在鉴定菌株水平的细菌,这在现代微生物学中变得越来越重要,有助于了解病原体传播途径和解决感染源问题。本研究旨在为产气荚膜梭菌建立一个公开的全基因组多位点序列分型(MLST)方案。总共选择了 1431 个高度保守的核心基因(1.34 兆碱基;参考基因组基因的 50%),用于产气荚膜梭菌基于核心基因组的多位点序列分型(cgMLST)方案。该方案应用于 282 个生态和地理多样化的基因组,结果表明,cgMLST 的基因分型结果在分辨率和树拓扑结构方面与基于核心基因组的单核苷酸多态性分型高度一致。此外,cgMLST 比经典 MLST 方法对产气荚膜梭菌具有更高的分辨率。通过重新研究美国和英国发表的产气荚膜梭菌相关感染暴发,证实了该方案用于暴发分析的可用性。总之,已经建立了一个公开的方案和一个用于产气荚膜梭菌基因组分型的等位基因命名法数据库,可用于广泛和标准化的流行病学研究。全球细菌病原体的流行病学监测因标准工具的可用性和分型数据的共享而得到加强。全基因组测序的使用为细菌菌株的高分辨率特征分析提供了可能,甚至可以达到克隆和亚克隆水平。核心基因组多位点序列分型是一种使用高度保守的核心基因进行深度基因分型的稳健系统。该方法已成功并广泛用于描述各种细菌物种的流行病学。然而,目前产气荚膜梭菌的 cgMLST 分型方案尚不可用。在本研究中,我们(i)开发了产气荚膜梭菌的 cgMLST 分型方案,(ii)评估了该方案在不同宿主和地理区域以及不同暴发情况下的产气荚膜梭菌基因组上的性能,最后,(iii)通过一个等位基因命名数据库为全球和标准的基因组分型提供支持,使该方案公开可用。