Elbrecht Vasco, Bourlat Sarah J, Hörren Thomas, Lindner Angie, Mordente Adriana, Noll Niklas W, Schäffler Livia, Sorg Martin, Zizka Vera M A
Centre for Biodiversity Monitoring, Zoological Research Museum Alexander Koenig, Bonn, Germany.
SimplexDNA AG, Winterthur, Switzerland.
PeerJ. 2021 Oct 5;9:e12177. doi: 10.7717/peerj.12177. eCollection 2021.
Small and rare specimens can remain undetected when metabarcoding is applied on bulk samples with a high specimen size heterogeneity. This is especially critical for Malaise trap samples, where most of the biodiversity is contributed by small taxa with low biomass. The separation of samples in different size fractions for downstream analysis is one possibility to increase detection of small and rare taxa. However, experiments systematically testing different size sorting approaches and subsequent proportional pooling of fractions are lacking, but would provide important information for the optimization of metabarcoding protocols. We set out to find a size sorting strategy for Malaise trap samples that maximizes taxonomic recovery but remains scalable and time efficient.
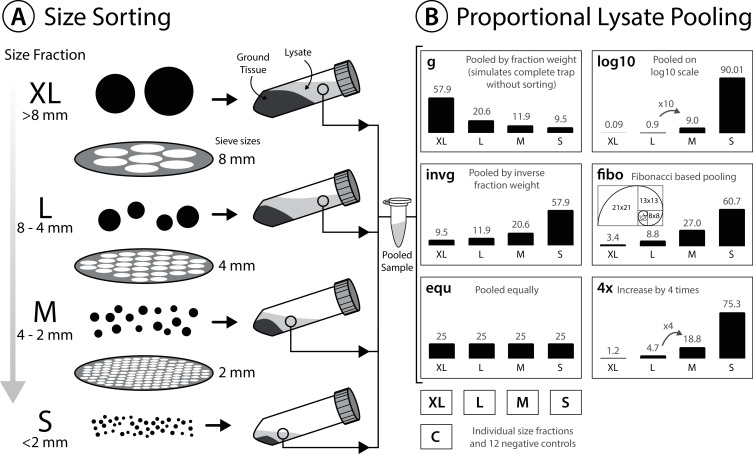
Three Malaise trap samples were sorted into four size classes using dry sieving. Each fraction was homogenized and lysed. The corresponding lysates were pooled to simulate unsorted samples. Pooling was additionally conducted in equal proportions and in four different proportions enriching the small size fraction of samples. DNA from the individual size classes as well as the pooled fractions was extracted and metabarcoded using the FwhF2 and Fol-degen-rev primer set. Additionally, alternative wet sieving strategies were explored.
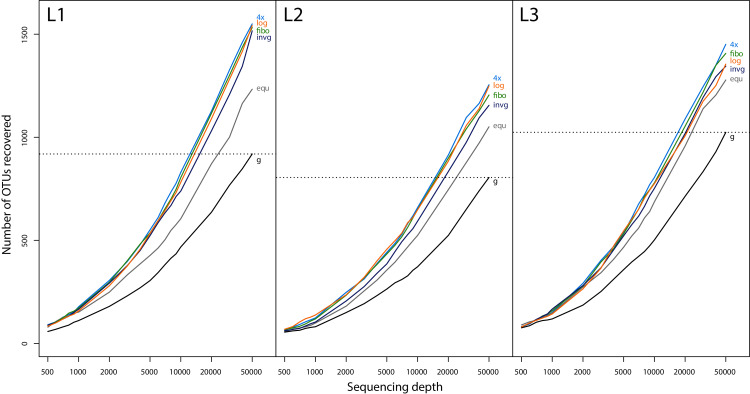
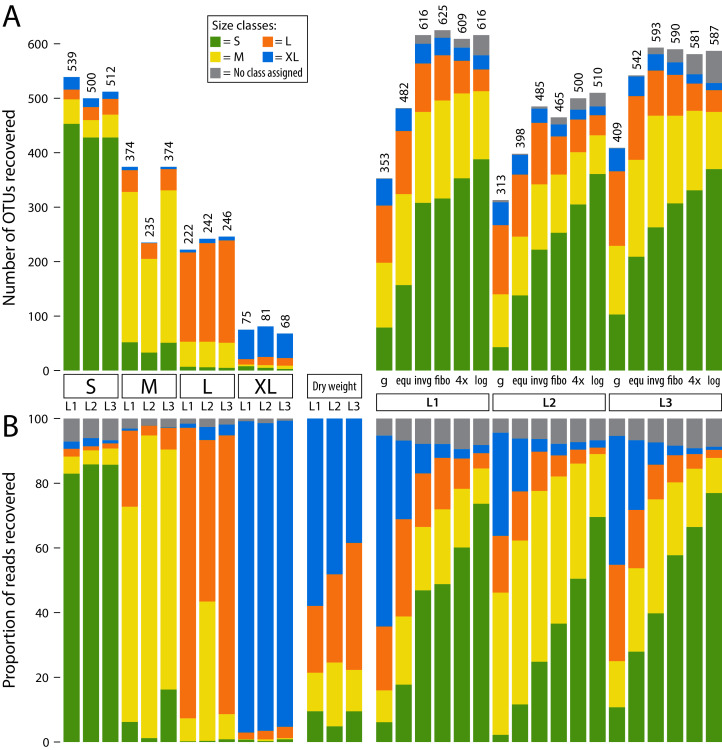
The small size fractions harboured the highest diversity and were best represented when pooling in favour of small specimens. Metabarcoding of unsorted samples decreases taxon recovery compared to size sorted samples. A size separation into only two fractions (below 4 mm and above) can double taxon recovery compared to not size sorting. However, increasing the sequencing depth 3- to 4-fold can also increase taxon recovery to levels comparable with size sorting, but remains biased towards biomass rich taxa in the sample.
We demonstrate that size fractionation of Malaise trap bulk samples can increase taxon recovery. While results show distinct patterns, the lack of statistical support due to the limited number of samples processed is a limitation. Due to increased speed and lower risk of cross-contamination as well as specimen damage we recommend wet sieving and proportional pooling of the lysates in favour of the small size fraction (80-90% volume). However, for large-scale projects with time constraints, increasing sequencing depth is an alternative solution.
当对具有高度样本大小异质性的大量样本应用宏条形码技术时,小型和稀有样本可能会未被检测到。这对于马氏网样本尤为关键,其中大部分生物多样性是由生物量低的小型分类群贡献的。将样本分离成不同大小的组分用于下游分析是增加小型和稀有分类群检测的一种可能性。然而,缺乏系统测试不同大小分选方法以及随后对各组分进行比例混合的实验,但这些实验将为宏条形码技术方案的优化提供重要信息。我们着手寻找一种适用于马氏网样本的大小分选策略,该策略能使分类群回收率最大化,同时保持可扩展性和时间效率。
使用干筛法将三个马氏网样本分选成四个大小类别。对每个组分进行匀浆和裂解。将相应的裂解物混合以模拟未分选的样本。还以等比例以及四种不同比例进行混合,以富集样本的小尺寸组分。提取各个大小类别的DNA以及混合后的组分,并使用FwhF2和Fol-degen-rev引物组进行宏条形码分析。此外,还探索了替代的湿筛策略。
小尺寸组分具有最高的多样性,并且在有利于小样本的混合时表现最佳。与大小分选后的样本相比,未分选样本的宏条形码分析降低了分类群回收率。与不进行大小分选相比,仅将样本分成两个组分(4毫米以下和以上)可使分类群回收率提高一倍。然而,将测序深度增加3至4倍也可使分类群回收率提高到与大小分选相当的水平,但仍偏向于样本中生物量丰富的分类群。
我们证明了马氏网大量样本的大小分级可以提高分类群回收率。虽然结果显示出明显的模式,但由于处理的样本数量有限而缺乏统计支持是一个限制因素。由于速度提高、交叉污染风险降低以及样本损伤减少,我们建议采用湿筛法并按比例混合裂解物以有利于小尺寸组分(80 - 90%体积)。然而,对于有时间限制的大型项目,增加测序深度是一种替代解决方案。