Khalak Y, Tresadern G, Aldeghi M, Baumann H M, Mobley D L, de Groot B L, Gapsys V
Computational Biomolecular Dynamics Group, Department of Theoretical and Computational Biophysics, Max Planck Institute for Biophysical Chemistry D-37077 Göttingen Germany
Computational Chemistry, Janssen Research & Development, Janssen Pharmaceutica N. V. Turnhoutseweg 30 2340 Beerse Belgium.
Chem Sci. 2021 Sep 24;12(41):13958-13971. doi: 10.1039/d1sc03472c. eCollection 2021 Oct 27.
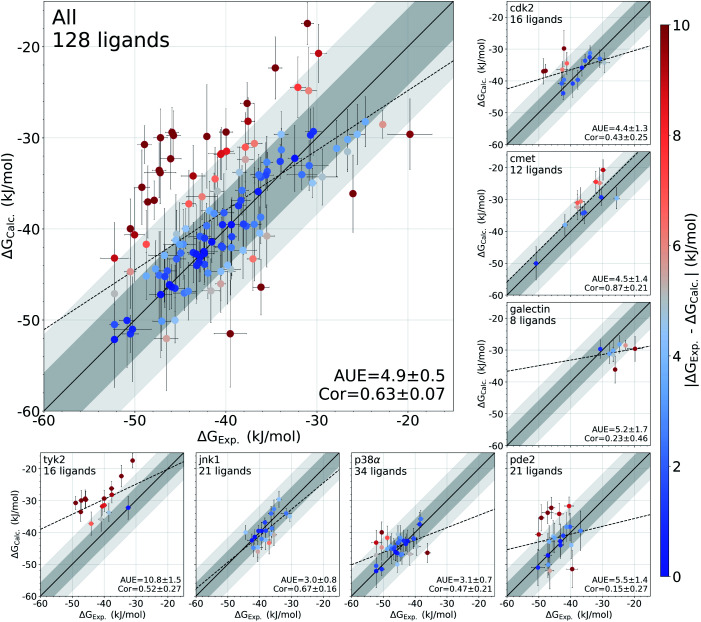
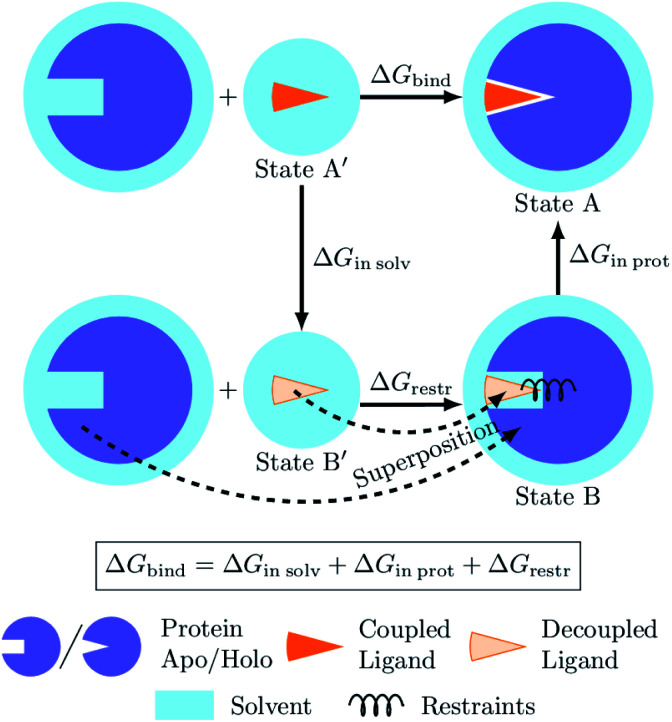
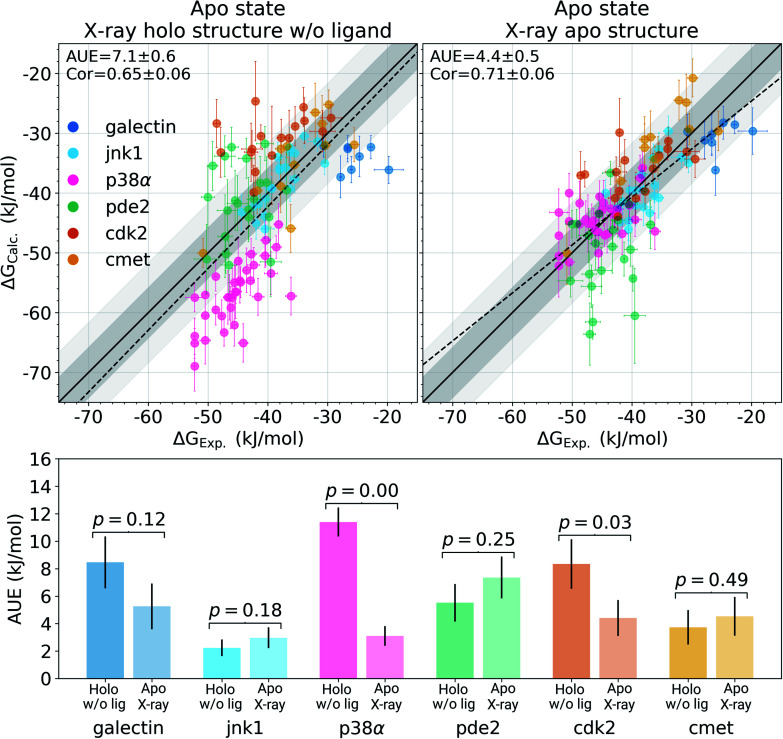
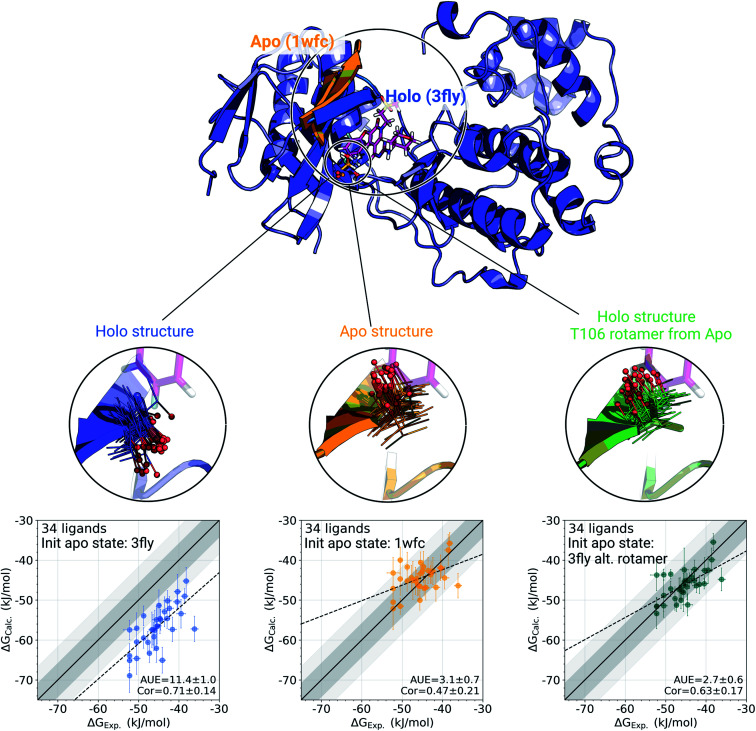
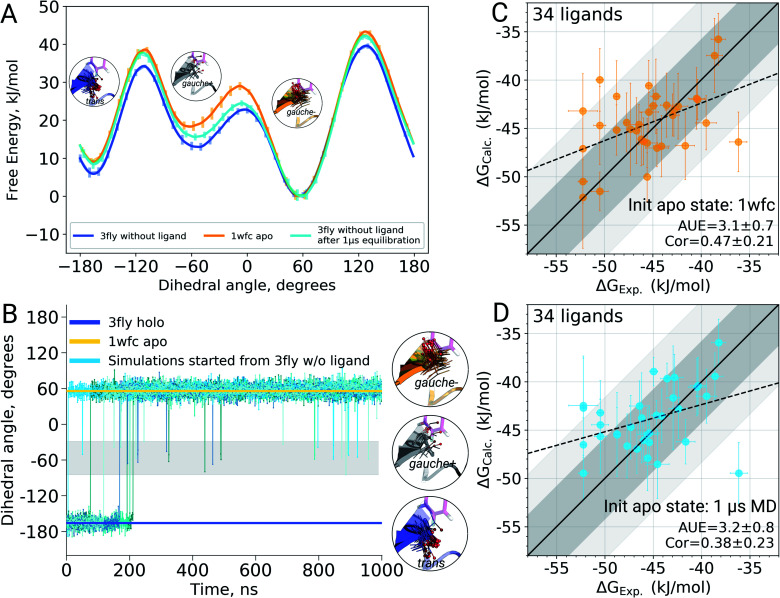
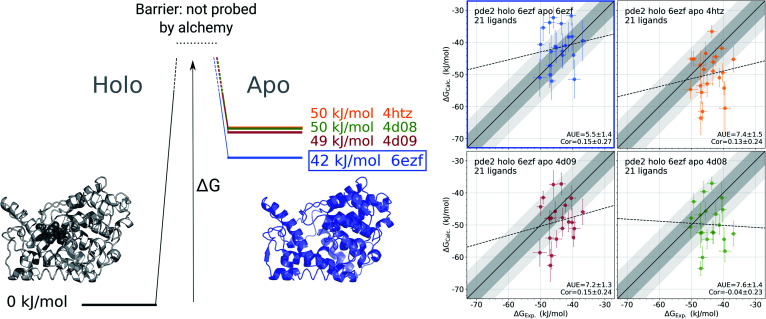
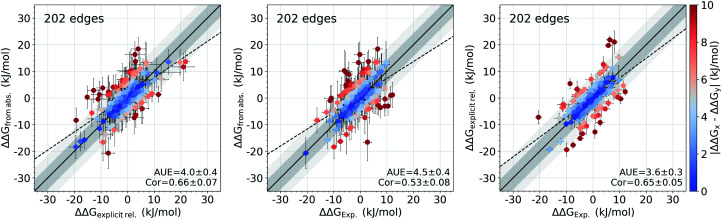
The recent advances in relative protein-ligand binding free energy calculations have shown the value of alchemical methods in drug discovery. Accurately assessing absolute binding free energies, although highly desired, remains a challenging endeavour, mostly limited to small model cases. Here, we demonstrate accurate first principles based absolute binding free energy estimates for 128 pharmaceutically relevant targets. We use a novel rigorous method to generate protein-ligand ensembles for the ligand in its decoupled state. Not only do the calculations deliver accurate protein-ligand binding affinity estimates, but they also provide detailed physical insight into the structural determinants of binding. We identify subtle rotamer rearrangements between apo and holo states of a protein that are crucial for binding. When compared to relative binding free energy calculations, obtaining absolute binding free energies is considerably more challenging in large part due to the need to explicitly account for the protein in its apo state. In this work we present several approaches to obtain apo state ensembles for accurate absolute Δ calculations, thus outlining protocols for prospective application of the methods for drug discovery.
相对蛋白质-配体结合自由能计算的最新进展已显示出炼金术方法在药物发现中的价值。准确评估绝对结合自由能,尽管非常令人期待,但仍然是一项具有挑战性的工作,主要限于小型模型案例。在此,我们展示了基于第一性原理对128个药物相关靶点进行的准确绝对结合自由能估计。我们使用一种新颖的严格方法来生成处于解耦状态的配体的蛋白质-配体集合。这些计算不仅能给出准确的蛋白质-配体结合亲和力估计值,还能提供对结合结构决定因素的详细物理洞察。我们识别出蛋白质的无配体状态和结合配体状态之间细微的旋转异构体重排,这些重排对于结合至关重要。与相对结合自由能计算相比,获得绝对结合自由能在很大程度上更具挑战性,这主要是因为需要明确考虑蛋白质的无配体状态。在这项工作中,我们提出了几种获得无配体状态集合以进行准确绝对Δ计算的方法,从而概述了这些方法在药物发现中的前瞻性应用方案。