Gapsys Vytautas, Pérez-Benito Laura, Aldeghi Matteo, Seeliger Daniel, van Vlijmen Herman, Tresadern Gary, de Groot Bert L
Computational Biomolecular Dynamics Group, Department of Theoretical and Computational Biophysics, Max Planck Institute for Biophysical Chemistry D-37077 Göttingen Germany
Computational Chemistry, Janssen Research & Development, Janssen Pharmaceutica N. V. Turnhoutseweg 30 B-2340 Beerse Belgium
Chem Sci. 2019 Dec 2;11(4):1140-1152. doi: 10.1039/c9sc03754c.
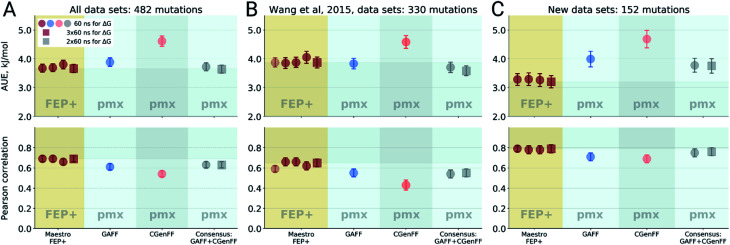
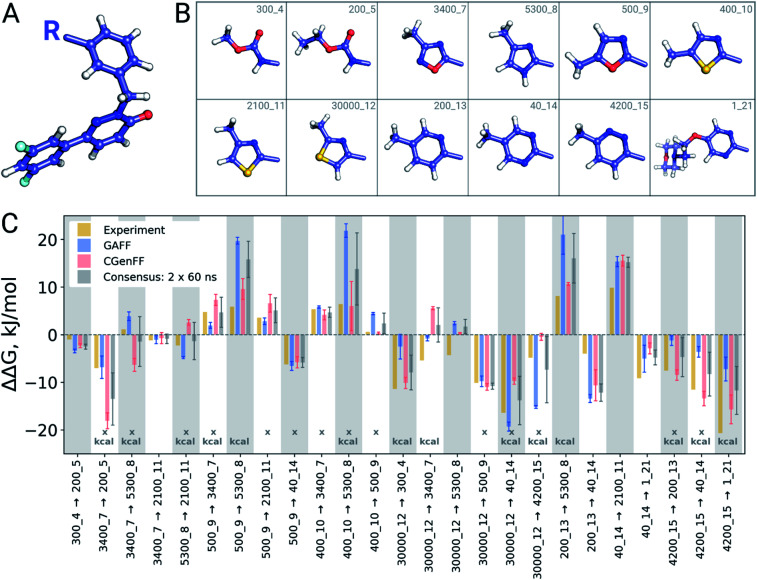
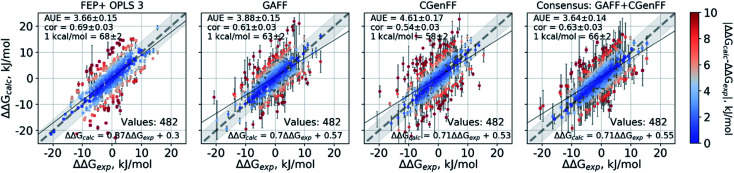
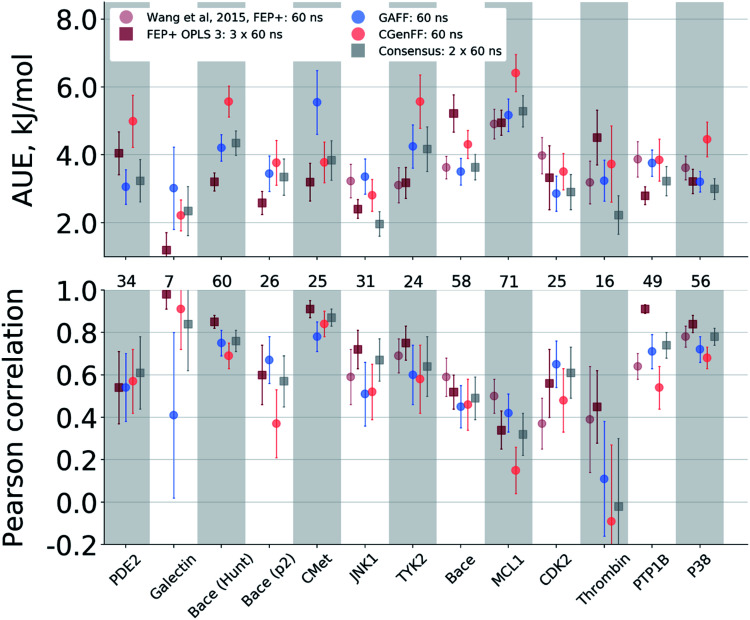
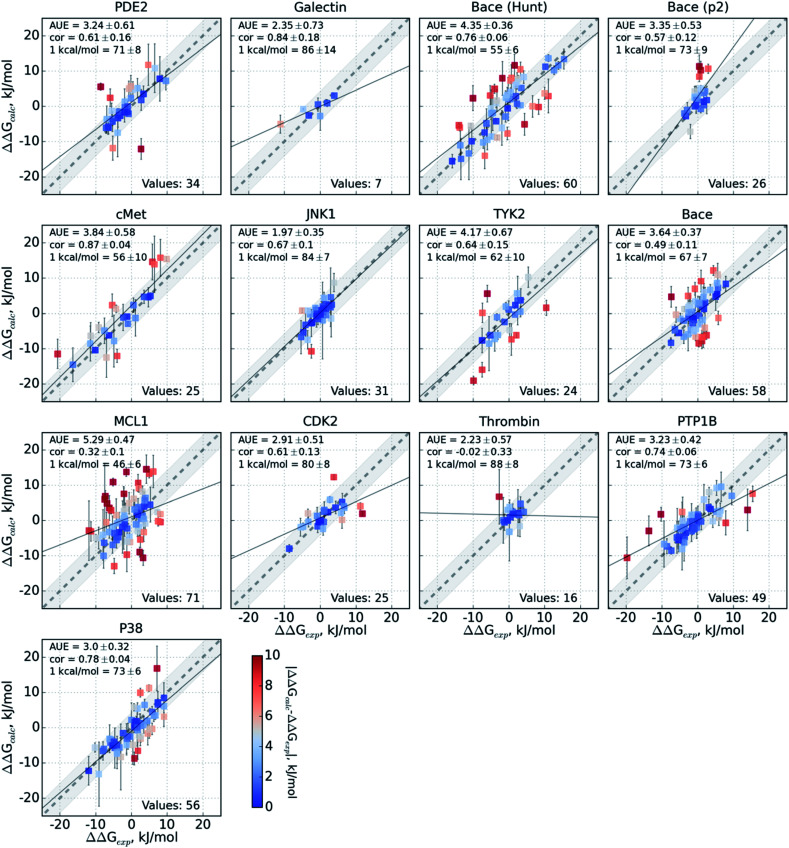
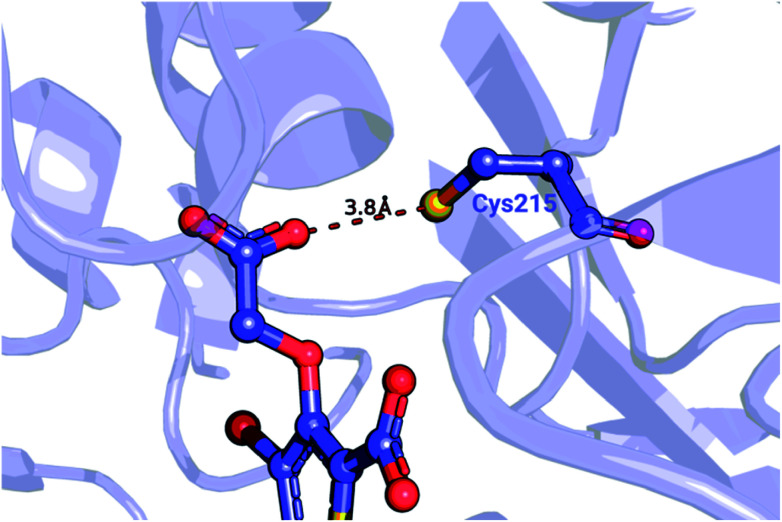
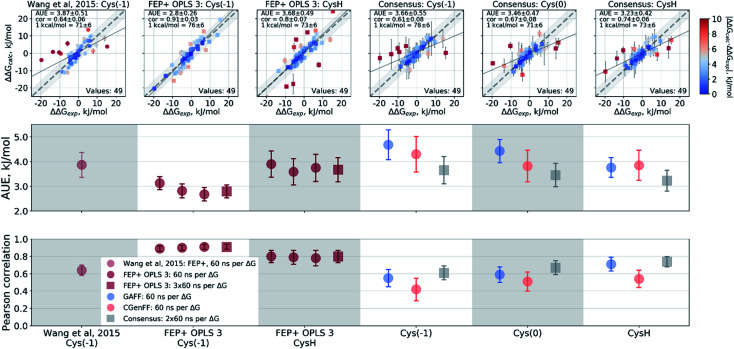
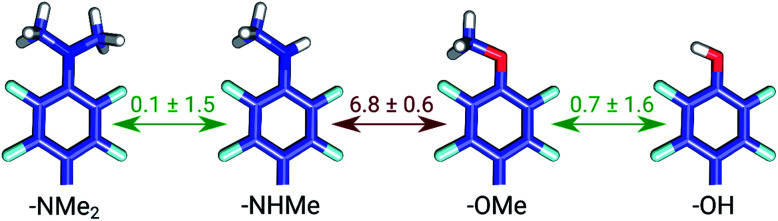
Ligand binding affinity calculations based on molecular dynamics (MD) simulations and non-physical (alchemical) thermodynamic cycles have shown great promise for structure-based drug design. However, their broad uptake and impact is held back by the notoriously complex setup of the calculations. Only a few tools other than the free energy perturbation approach by Schrödinger Inc. (referred to as FEP+) currently enable end-to-end application. Here, we present for the first time an approach based on the open-source software pmx that allows to easily set up and run alchemical calculations for diverse sets of small molecules using the GROMACS MD engine. The method relies on theoretically rigorous non-equilibrium thermodynamic integration (TI) foundations, and its flexibility allows calculations with multiple force fields. In this study, results from the Amber and Charmm force fields were combined to yield a consensus outcome performing on par with the commercial FEP+ approach. A large dataset of 482 perturbations from 13 different protein-ligand datasets led to an average unsigned error (AUE) of 3.64 ± 0.14 kJ mol, equivalent to Schrödinger's FEP+ AUE of 3.66 ± 0.14 kJ mol. For the first time, a setup is presented for overall high precision and high accuracy relative protein-ligand alchemical free energy calculations based on open-source software.
基于分子动力学(MD)模拟和非物理(炼金术)热力学循环的配体结合亲和力计算在基于结构的药物设计方面显示出巨大潜力。然而,计算设置极其复杂,这阻碍了它们的广泛应用和影响。目前,除了薛定谔公司的自由能微扰方法(称为FEP+)外,只有少数工具能够实现端到端应用。在此,我们首次提出一种基于开源软件pmx的方法,该方法允许使用GROMACS MD引擎轻松设置并运行针对各种小分子集的炼金术计算。该方法基于理论严谨的非平衡热力学积分(TI)基础,其灵活性允许使用多种力场进行计算。在本研究中,结合了Amber和Charmm力场的结果,以产生与商业FEP+方法相当的一致结果。来自13个不同蛋白质-配体数据集的482个微扰的大型数据集导致平均无符号误差(AUE)为3.64±0.14 kJ/mol,相当于薛定谔的FEP+的AUE为3.66±0.14 kJ/mol。首次提出了一种基于开源软件的、用于相对蛋白质-配体炼金术自由能计算的总体高精度和高准确性的设置。