ESAT-STADIUS, KU Leuven, 3001 Leuven, Belgium.
Institut Jean-Pierre Bourgin, Université Paris-Saclay, INRAE, AgroParisTech, 78000 Versailles, France.
Nucleic Acids Res. 2022 Feb 22;50(3):e16. doi: 10.1093/nar/gkab1099.
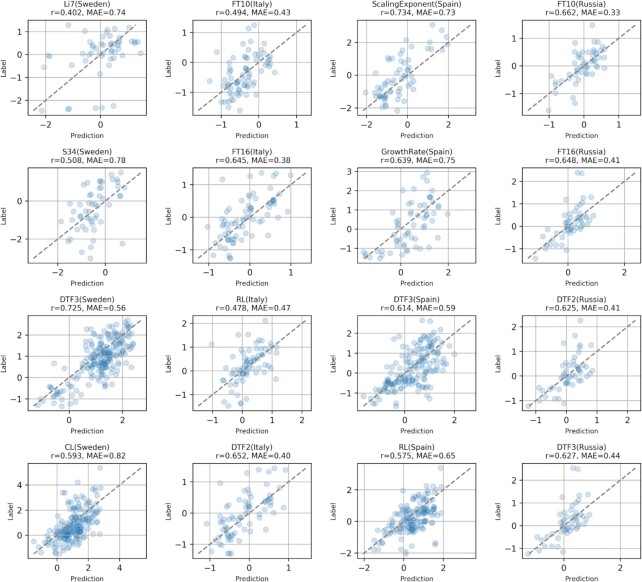
In many cases, the unprecedented availability of data provided by high-throughput sequencing has shifted the bottleneck from a data availability issue to a data interpretation issue, thus delaying the promised breakthroughs in genetics and precision medicine, for what concerns Human genetics, and phenotype prediction to improve plant adaptation to climate change and resistance to bioagressors, for what concerns plant sciences. In this paper, we propose a novel Genome Interpretation paradigm, which aims at directly modeling the genotype-to-phenotype relationship, and we focus on A. thaliana since it is the best studied model organism in plant genetics. Our model, called Galiana, is the first end-to-end Neural Network (NN) approach following the genomes in/phenotypes out paradigm and it is trained to predict 288 real-valued Arabidopsis thaliana phenotypes from Whole Genome sequencing data. We show that 75 of these phenotypes are predicted with a Pearson correlation ≥0.4, and are mostly related to flowering traits. We show that our end-to-end NN approach achieves better performances and larger phenotype coverage than models predicting single phenotypes from the GWAS-derived known associated genes. Galiana is also fully interpretable, thanks to the Saliency Maps gradient-based approaches. We followed this interpretation approach to identify 36 novel genes that are likely to be associated with flowering traits, finding evidence for 6 of them in the existing literature.
在许多情况下,高通量测序提供的前所未有的数据可用性已经将瓶颈从数据可用性问题转移到数据解释问题,从而延迟了遗传学和精准医学方面的承诺突破,就人类遗传学而言,以及表型预测以提高植物对气候变化的适应能力和对生物侵害者的抵抗力,就植物科学而言。在本文中,我们提出了一种新的基因组解释范例,旨在直接模拟基因型到表型的关系,我们专注于拟南芥,因为它是植物遗传学中研究最好的模式生物。我们的模型称为 Galiana,是第一个遵循基因组内/表型外范例的端到端神经网络 (NN) 方法,它经过训练可从全基因组测序数据中预测 288 个真实的拟南芥表型。我们表明,其中 75 个表型的 Pearson 相关系数≥0.4,且主要与开花性状有关。我们表明,我们的端到端神经网络方法比从 GWAS 衍生的已知相关基因预测单个表型的模型具有更好的性能和更大的表型覆盖范围。Galiana 也具有完全可解释性,这要归功于基于梯度的显着性图方法。我们遵循这种解释方法来识别 36 个可能与开花性状相关的新基因,并在现有文献中找到了其中 6 个的证据。