Zhang Mengge, Yang Lianping, Ren Jie, Ahlgren Nathan A, Fuhrman Jed A, Sun Fengzhu
Molecular and Computational Biology Program, University of Southern California, Los Angeles, California, USA.
College of Sciences, Northeastern University, Shenyang, China.
BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):60. doi: 10.1186/s12859-017-1473-7.
The study of virus-host infectious association is important for understanding the functions and dynamics of microbial communities. Both cellular and fractionated viral metagenomic data generate a large number of viral contigs with missing host information. Although relative simple methods based on the similarity between the word frequency vectors of viruses and bacterial hosts have been developed to study virus-host associations, the problem is significantly understudied. We hypothesize that machine learning methods based on word frequencies can be efficiently used to study virus-host infectious associations.
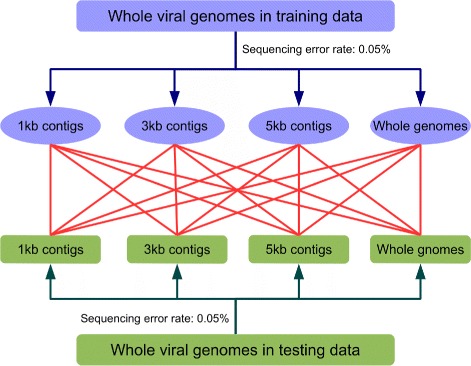
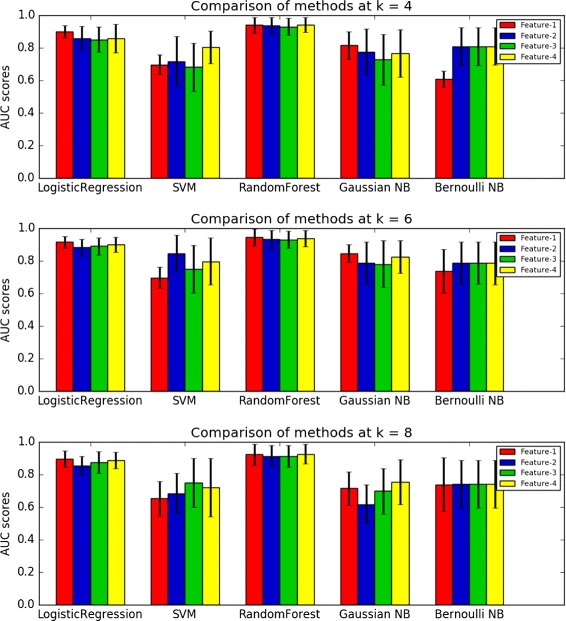
We investigate four different representations of word frequencies of viral sequences including the relative word frequency and three normalized word frequencies by subtracting the number of expected from the observed word counts. We also study five machine learning methods including logistic regression, support vector machine, random forest, Gaussian naive Bayes and Bernoulli naive Bayes for separating infectious from non-infectious viruses for nine bacterial host genera with at least 45 infecting viruses. Area under the receiver operating characteristic curve (AUC) is used to compare the performance of different machine learning method and feature combinations. We then evaluate the performance of the best method for the identification of the hosts of contigs in metagenomic studies. We also develop a maximum likelihood method to estimate the fraction of true infectious viruses for a given host in viral tagging experiments.
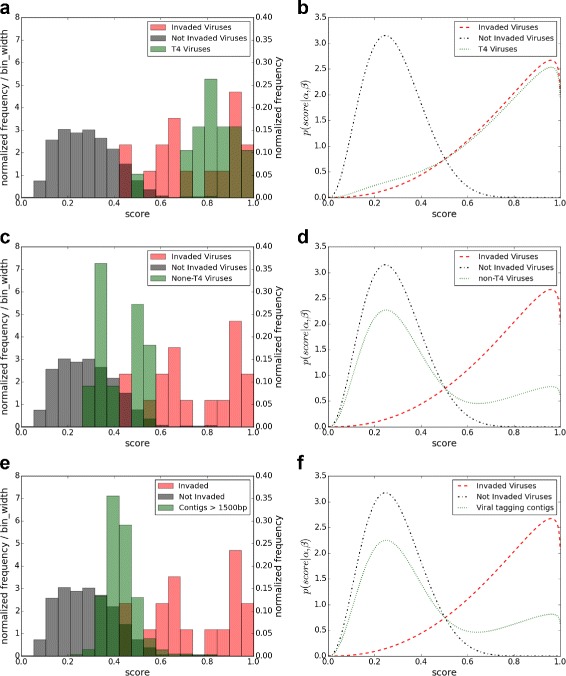
Based on nine bacterial host genera with at least 45 infectious viruses, we show that random forest together with the relative word frequency vector performs the best in identifying viruses infecting particular hosts. For all the nine host genera, the AUC is over 0.85 and for five of them, the AUC is higher than 0.98 when the word size is 6 indicating the high accuracy of using machine learning approaches for identifying viruses infecting particular hosts. We also show that our method can predict the hosts of viral contigs of length at least 1kbps in metagenomic studies with high accuracy. The random forest together with word frequency vector outperforms current available methods based on Manhattan and [Formula: see text] dissimilarity measures. Based on word frequencies, we estimate that about 95% of the identified T4-like viruses in viral tagging experiment infect Synechococcus, while only about 29% of the identified non-T4-like viruses and 30% of the contigs in the study potentially infect Synechococcus.
The random forest machine learning method together with the relative word frequencies as features of viruses can be used to predict viruses and viral contigs for specific bacterial hosts. The maximum likelihood approach can be used to estimate the fraction of true infectious associated viruses in viral tagging experiments.
病毒-宿主感染关联的研究对于理解微生物群落的功能和动态至关重要。细胞和分级病毒宏基因组数据都会产生大量缺少宿主信息的病毒重叠群。尽管已经开发了基于病毒和细菌宿主词频向量之间相似性的相对简单的方法来研究病毒-宿主关联,但这个问题仍未得到充分研究。我们假设基于词频的机器学习方法可以有效地用于研究病毒-宿主感染关联。
我们研究了病毒序列词频的四种不同表示形式,包括相对词频以及通过从观察到的词计数中减去预期数量得到的三种归一化词频。我们还研究了五种机器学习方法,包括逻辑回归、支持向量机、随机森林、高斯朴素贝叶斯和伯努利朴素贝叶斯,用于区分九种细菌宿主属的感染性病毒和非感染性病毒,每种宿主属至少有45种感染病毒。使用受试者工作特征曲线下面积(AUC)来比较不同机器学习方法和特征组合的性能。然后,我们评估最佳方法在宏基因组研究中识别重叠群宿主的性能。我们还开发了一种最大似然方法来估计病毒标记实验中给定宿主的真正感染性病毒的比例。
基于九种细菌宿主属,每种宿主属至少有45种感染性病毒,我们表明随机森林与相对词频向量相结合在识别感染特定宿主的病毒方面表现最佳。对于所有九种宿主属,当词大小为6时,AUC超过0.85,其中五种宿主属的AUC高于0.98,这表明使用机器学习方法识别感染特定宿主的病毒具有很高的准确性。我们还表明,我们的方法可以在宏基因组研究中高精度地预测长度至少为1kbps的病毒重叠群的宿主。随机森林与词频向量相结合的方法优于目前基于曼哈顿和[公式:见原文]差异度量的可用方法。基于词频,我们估计在病毒标记实验中鉴定出的约95%的类T4病毒感染了聚球藻属,而在该研究中鉴定出的非类T4病毒中只有约29%以及重叠群中只有30%可能感染聚球藻属。
随机森林机器学习方法与作为病毒特征的相对词频相结合,可用于预测特定细菌宿主的病毒和病毒重叠群。最大似然方法可用于估计病毒标记实验中真正感染相关病毒的比例。