Division of Plant Pathology, ICAR-Indian Agricultural Research Institute, New Delhi, 110012, India.
Crop Improvement Group, International Centre for Genetic Engineering and Biotechnology, New Delhi, 110067, India.
Sci Rep. 2021 Nov 25;11(1):22922. doi: 10.1038/s41598-021-01980-2.
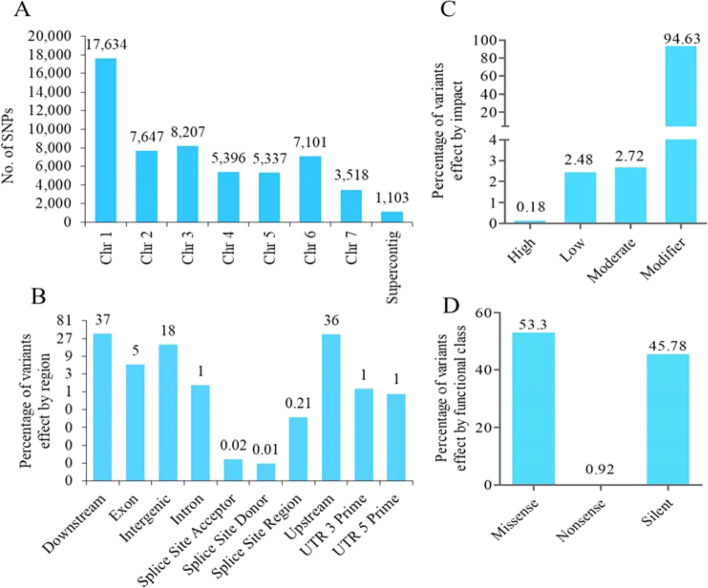
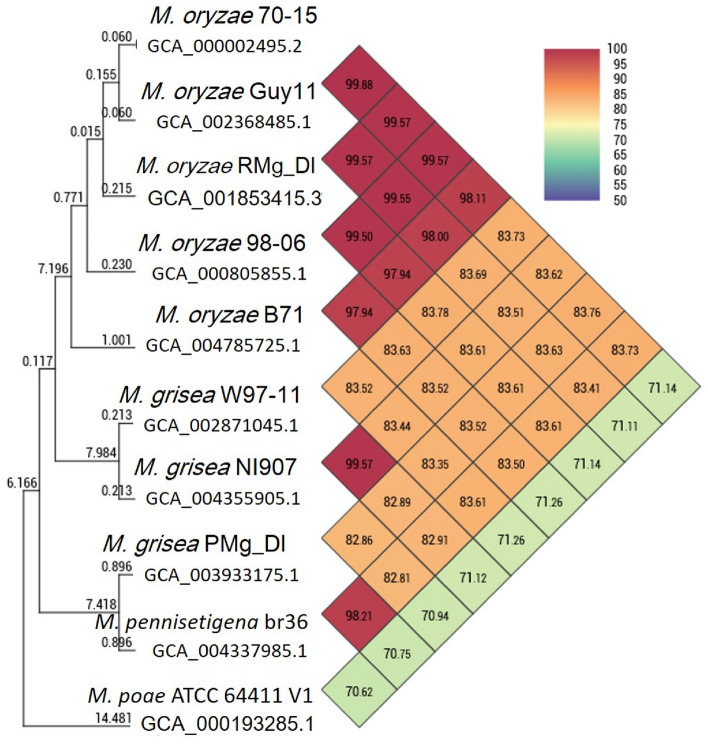
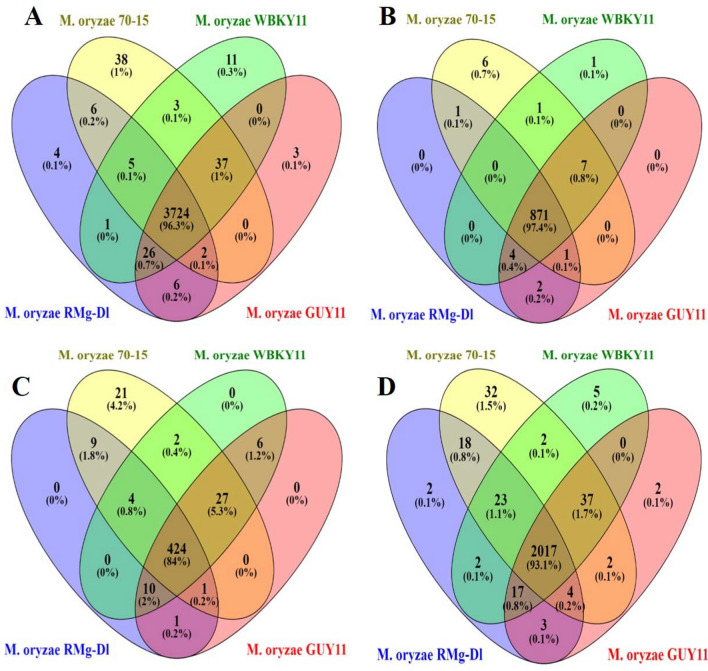
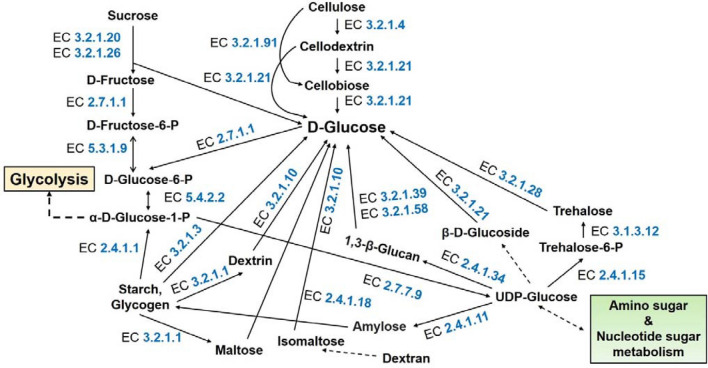
Blast disease incited by Magnaporthe oryzae is a major threat to sustain rice production in all rice growing nations. The pathogen is widely distributed in all rice paddies and displays rapid aerial transmissions, and seed-borne latent infection. In order to understand the genetic variability, host specificity, and molecular basis of the pathogenicity-associated traits, the whole genome of rice infecting Magnaporthe oryzae (Strain RMg_Dl) was sequenced using the Illumina and PacBio (RSII compatible) platforms. The high-throughput hybrid assembly of short and long reads resulted in a total of 375 scaffolds with a genome size of 42.43 Mb. Furthermore, comparative genome analysis revealed 99% average nucleotide identity (ANI) with other oryzae genomes and 83% against M. grisea, and 73% against M. poe genomes. The gene calling identified 10,553 genes with 10,539 protein-coding sequences. Among the detected transposable elements, the LTR/Gypsy and Type LINE showed high occurrence. The InterProScan of predicted protein sequences revealed that 97% protein family (PFAM), 98% superfamily, and 95% CDD were shared among RMg_Dl and reference 70-15 genome, respectively. Additionally, 550 CAZymes with high GH family content/distribution and cell wall degrading enzymes (CWDE) such endoglucanase, beta-glucosidase, and pectate lyase were also deciphered in RMg_Dl. The prevalence of virulence factors determination revealed that 51 different VFs were found in the genome. The biochemical pathway such as starch and sucrose metabolism, mTOR signaling, cAMP signaling, MAPK signaling pathways related genes were identified in the genome. The 49,065 SNPs, 3267 insertions and 3611 deletions were detected, and majority of these varinats were located on downstream and upstream region. Taken together, the generated information will be useful to develop a specific marker for diagnosis, pathogen surveillance and tracking, molecular taxonomy, and species delineation which ultimately leads to device improved management strategies for blast disease.
稻瘟病是由稻瘟病菌引起的,是所有种植水稻的国家维持水稻生产的主要威胁。该病原体广泛分布于所有稻田,并表现出快速的空气传播和种子传播的潜伏感染。为了了解与致病性相关的遗传变异性、宿主特异性和分子基础,使用 Illumina 和 PacBio(兼容 RSII)平台对感染水稻的稻瘟病菌(菌株 RMg_Dl)的全基因组进行了测序。高通量混合短读长和长读长组装产生了总共 375 个基因组支架,基因组大小为 42.43 Mb。此外,比较基因组分析显示与其他籼稻基因组的平均核苷酸同一性(ANI)为 99%,与稻瘟病菌的 ANI 为 83%,与稻曲病菌的 ANI 为 73%。基因预测鉴定出 10553 个基因,其中包括 10539 个蛋白编码序列。在所检测到的转座元件中,LTR/Gypsy 和 Type LINE 出现频率较高。预测蛋白序列的 InterProScan 显示,RMg_Dl 和参考 70-15 基因组分别有 97%的蛋白家族(PFAM)、98%的超家族和 95%的 CDD 共享。此外,还在 RMg_Dl 中破译了 550 种具有高 GH 家族含量/分布和细胞壁降解酶(CWDE)的 CAZymes,如内切葡聚糖酶、β-葡萄糖苷酶和果胶裂解酶。在毒力因子决定因素的普遍性测定中发现,在基因组中发现了 51 种不同的 VFs。在基因组中鉴定了与淀粉和蔗糖代谢、mTOR 信号、cAMP 信号、MAPK 信号通路相关的基因。检测到 49065 个 SNPs、3267 个插入和 3611 个缺失,这些变异的大部分位于下游和上游区域。总之,所产生的信息将有助于开发用于诊断、病原体监测和跟踪、分子分类学和物种划定的特定标记,最终导致制定改进的稻瘟病管理策略。