Shen Chunmei, Shen Ying, Zhang Hui, Xu Maosuo, He Leqi, Qie Jingbo
Department of Hospital Infection Management, The Fifth People's Hospital of Shanghai, Fudan University, Shanghai, China.
Department of Clinical Laboratory Medicine, The Fifth People's Hospital of Shanghai, Fudan University, Shanghai, China.
Front Microbiol. 2021 Nov 18;12:773829. doi: 10.3389/fmicb.2021.773829. eCollection 2021.
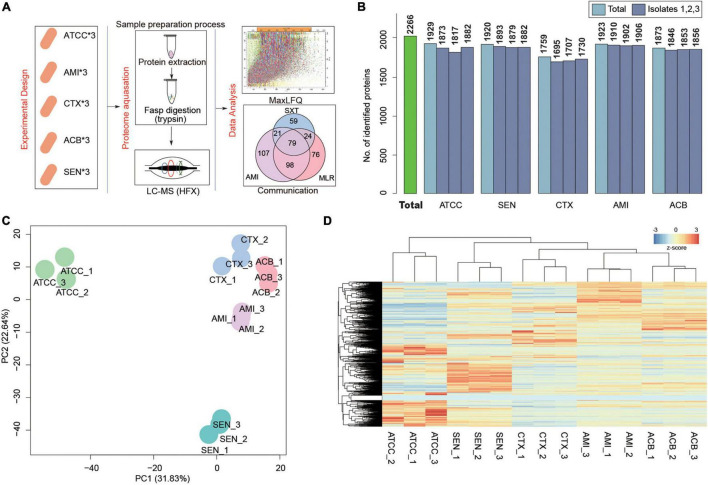
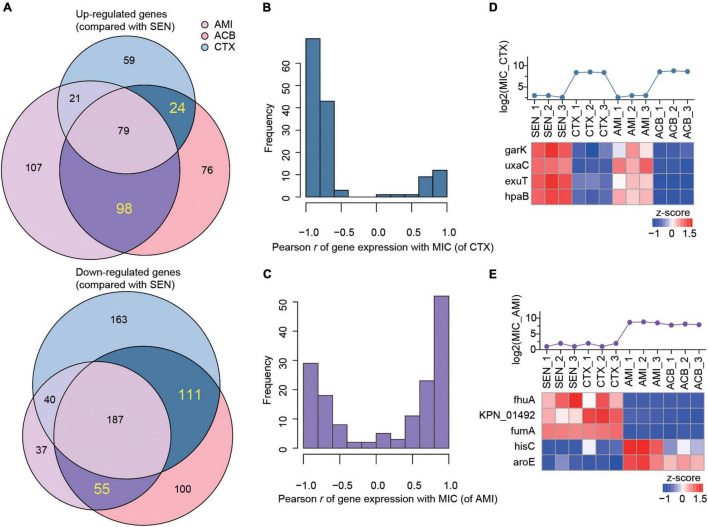
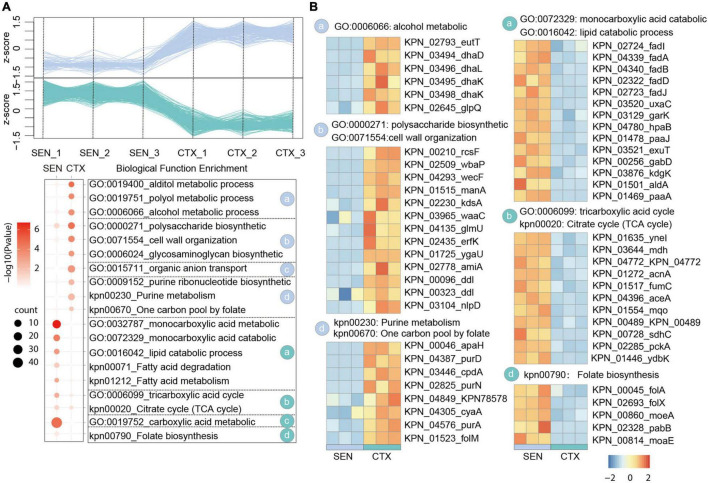
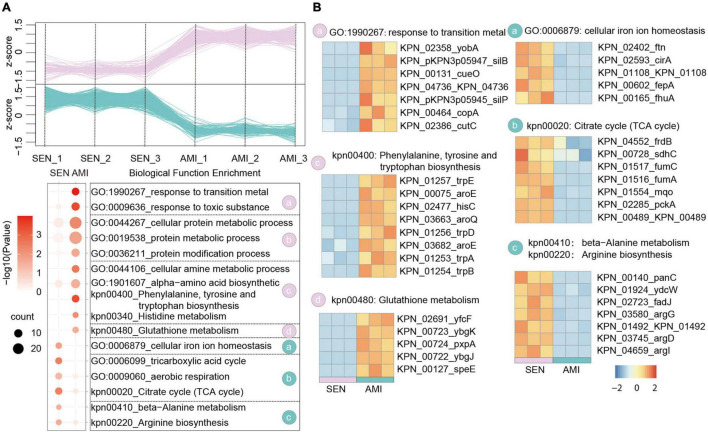
Antibiotic resistance (AMR) has always been a hot topic all over the world and its mechanisms are varied and complicated. Previous evidence revealed the metabolic slowdown in resistant bacteria, suggesting the important role of metabolism in antibiotic resistance. However, the molecular mechanism of reduced metabolism remains poorly understood, which inspires us to explore the global proteome change during antibiotic resistance. Here, the sensitive, cotrimoxazole-resistant, amikacin-resistant, and amikacin/cotrimoxazole -both-resistant KPN clinical isolates were collected and subjected to proteome analysis through liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). A deep coverage of 2,266 proteins were successfully identified and quantified in total, representing the most comprehensive protein quantification data by now. Further bioinformatic analysis showed down-regulation of tricarboxylic acid cycle (TCA) pathway and up-regulation of alcohol metabolic or glutathione metabolism processes, which may contribute to ROS clearance and cell survival, in drug-resistant isolates. These results indicated that metabolic pathway alteration was directly correlated with antibiotic resistance, which could promote the development of antibacterial drugs from "target" to "network." Moreover, combined with minimum inhibitory concentration (MIC) of cotrimoxazole and amikacin on different KPN isolates, we identified nine proteins, including garK, uxaC, exuT, hpaB, fhuA, KPN_01492, fumA, hisC, and aroE, which might contribute mostly to the survival of KPN under drug pressure. In sum, our findings provided novel, non-antibiotic-based therapeutics against resistant KPN.
抗生素耐药性(AMR)一直是全球的热门话题,其机制多样且复杂。先前的证据揭示了耐药细菌的代谢减缓,这表明代谢在抗生素耐药性中起着重要作用。然而,代谢减缓的分子机制仍知之甚少,这促使我们探索抗生素耐药过程中的全球蛋白质组变化。在此,收集了对敏感、耐复方新诺明、耐阿米卡星以及耐阿米卡星/复方新诺明的肺炎克雷伯菌临床分离株,并通过液相色谱-串联质谱(LC-MS/MS)进行蛋白质组分析。总共成功鉴定和定量了2266种蛋白质,这是目前最全面的蛋白质定量数据。进一步的生物信息学分析表明,耐药分离株中三羧酸循环(TCA)途径下调,而酒精代谢或谷胱甘肽代谢过程上调,这可能有助于活性氧清除和细胞存活。这些结果表明,代谢途径改变与抗生素耐药性直接相关,这可能推动抗菌药物从“靶点”向“网络”发展。此外,结合复方新诺明和阿米卡星对不同肺炎克雷伯菌分离株的最低抑菌浓度(MIC),我们鉴定出9种蛋白质,包括garK、uxaC、exuT、hpaB、fhuA、KPN_01492、fumA、hisC和aroE,它们可能对肺炎克雷伯菌在药物压力下的存活贡献最大。总之,我们的研究结果为抗耐药肺炎克雷伯菌提供了基于非抗生素的新型治疗方法。