Department of Physiology and Biophysics, Weill Cornell Medicine, New York, New York, USA.
The HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine, Weill Cornell Medicine, New York, New York, USA.
Genome Biol. 2021 Dec 6;22(1):332. doi: 10.1186/s13059-021-02529-2.
Cytosine modifications in DNA such as 5-methylcytosine (5mC) underlie a broad range of developmental processes, maintain cellular lineage specification, and can define or stratify types of cancer and other diseases. However, the wide variety of approaches available to interrogate these modifications has created a need for harmonized materials, methods, and rigorous benchmarking to improve genome-wide methylome sequencing applications in clinical and basic research. Here, we present a multi-platform assessment and cross-validated resource for epigenetics research from the FDA's Epigenomics Quality Control Group.
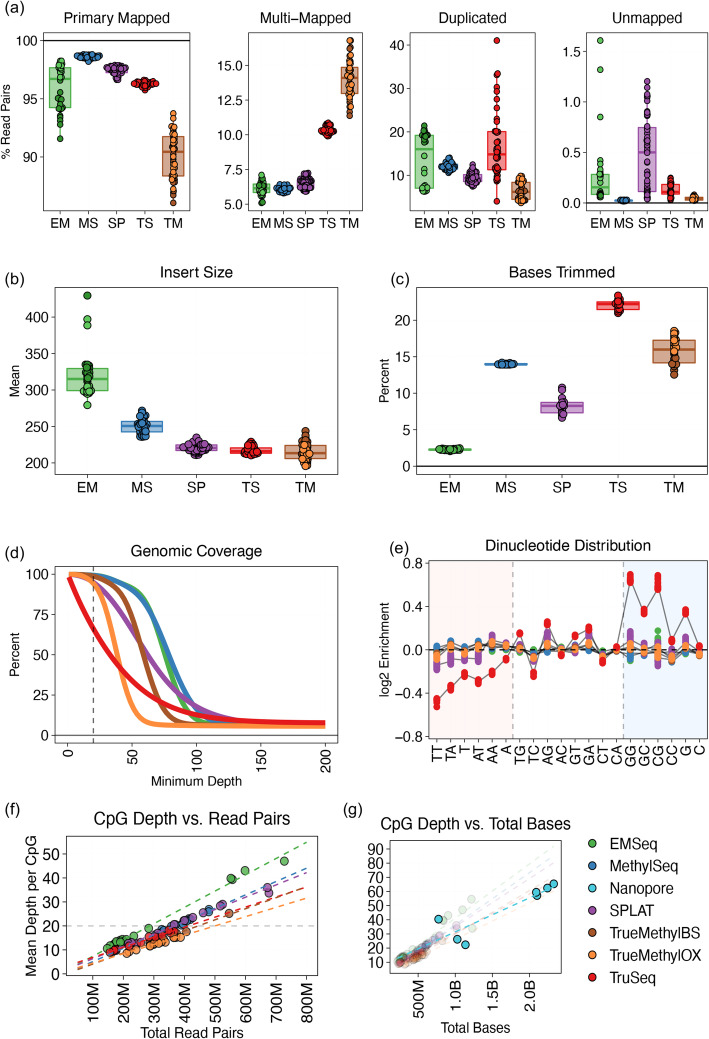
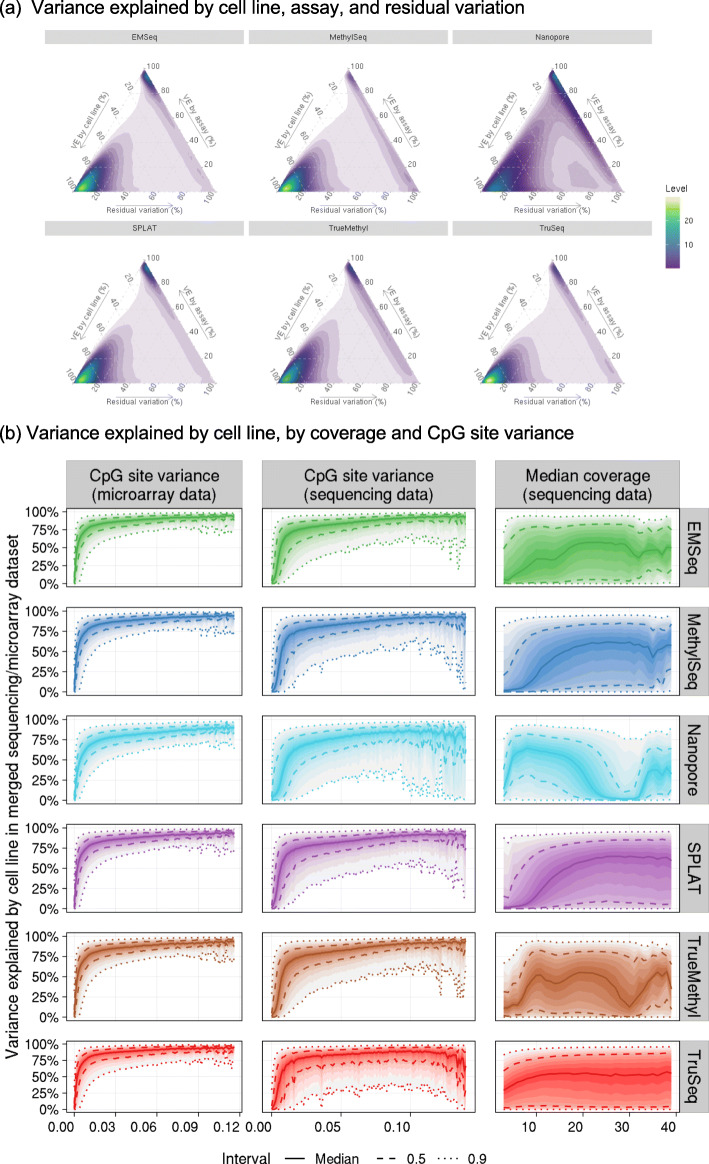
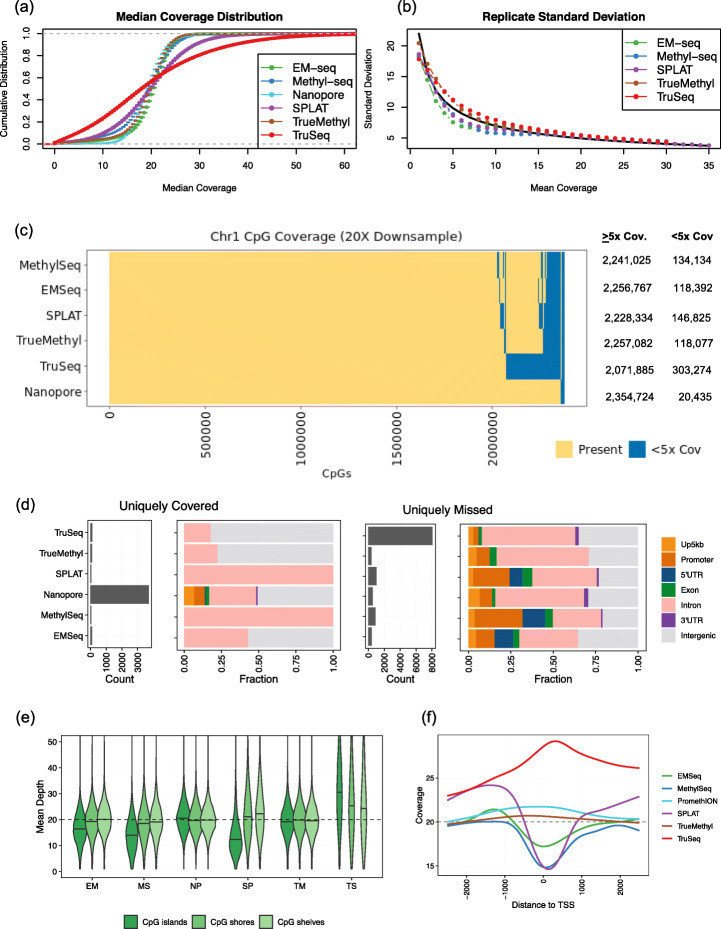
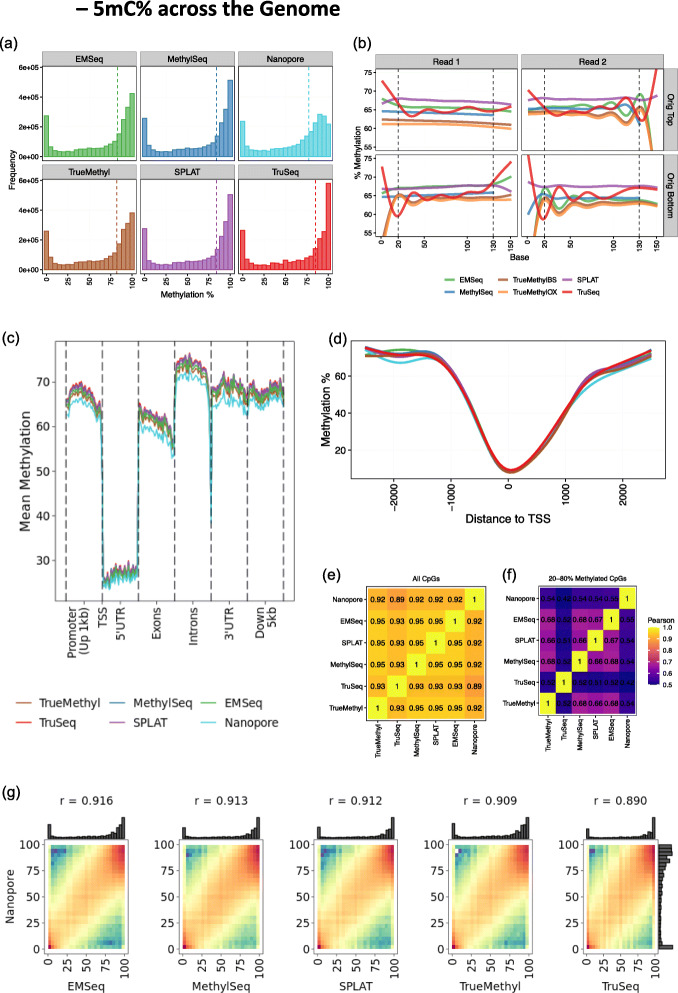
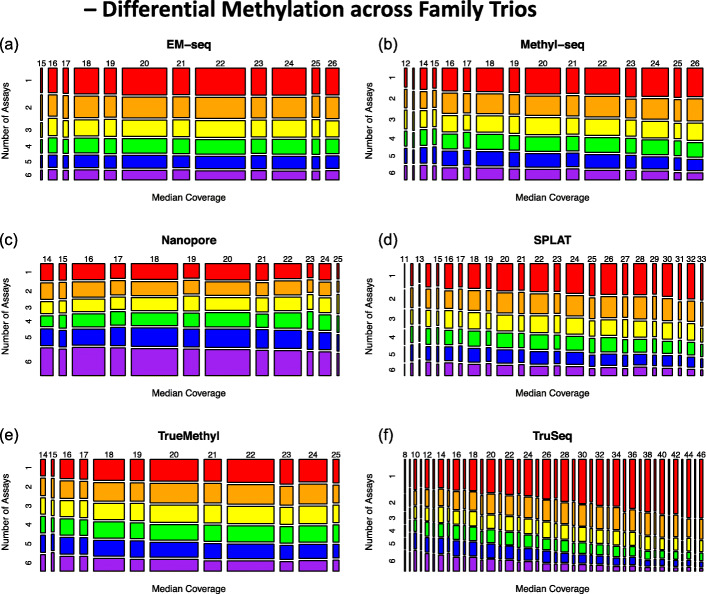
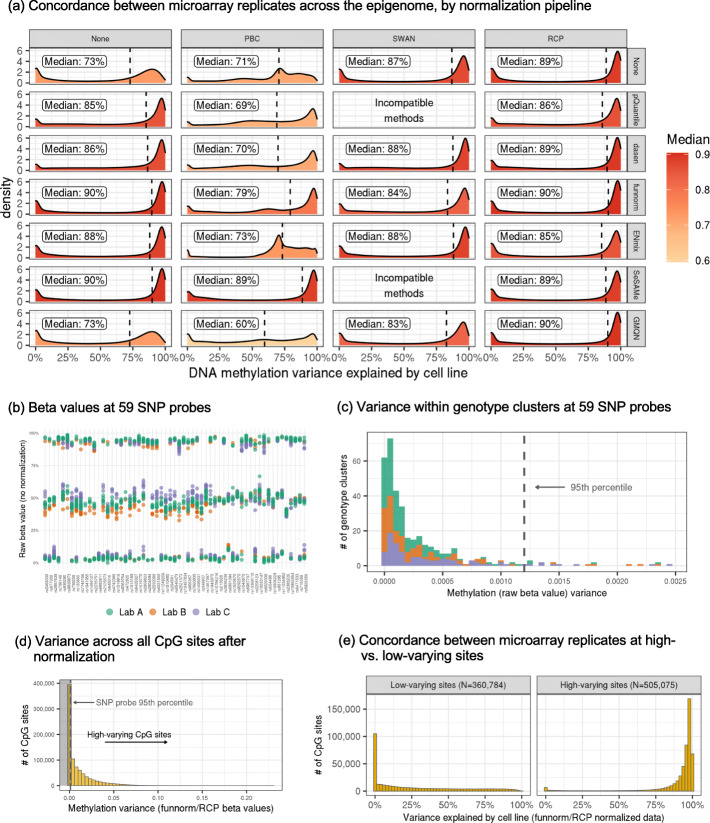
Each sample is processed in multiple replicates by three whole-genome bisulfite sequencing (WGBS) protocols (TruSeq DNA methylation, Accel-NGS MethylSeq, and SPLAT), oxidative bisulfite sequencing (TrueMethyl), enzymatic deamination method (EMSeq), targeted methylation sequencing (Illumina Methyl Capture EPIC), single-molecule long-read nanopore sequencing from Oxford Nanopore Technologies, and 850k Illumina methylation arrays. After rigorous quality assessment and comparison to Illumina EPIC methylation microarrays and testing on a range of algorithms (Bismark, BitmapperBS, bwa-meth, and BitMapperBS), we find overall high concordance between assays, but also differences in efficiency of read mapping, CpG capture, coverage, and platform performance, and variable performance across 26 microarray normalization algorithms.
The data provided herein can guide the use of these DNA reference materials in epigenomics research, as well as provide best practices for experimental design in future studies. By leveraging seven human cell lines that are designated as publicly available reference materials, these data can be used as a baseline to advance epigenomics research.
DNA 中的胞嘧啶修饰,如 5-甲基胞嘧啶(5mC),是广泛的发育过程的基础,维持细胞谱系的特异性,并可以定义或分层癌症和其他疾病的类型。然而,用于研究这些修饰的各种方法需要协调一致的材料、方法和严格的基准测试,以改善临床和基础研究中全基因组甲基组测序的应用。在这里,我们展示了来自 FDA 表观基因组学质量控制小组的多平台评估和交叉验证的表观遗传学研究资源。
每个样本通过三种全基因组亚硫酸氢盐测序(WGBS)方案(TruSeq DNA 甲基化、Accel-NGS MethylSeq 和 SPLAT)、氧化亚硫酸氢盐测序(TrueMethyl)、酶促脱氨方法(EMSeq)、靶向甲基化测序(Illumina Methyl Capture EPIC)、牛津纳米孔技术的单分子长读长纳米孔测序和 Illumina 850k 甲基化芯片进行多次重复处理。经过严格的质量评估,并与 Illumina EPIC 甲基化微阵列进行比较,并在一系列算法(Bismark、BitmapperBS、bwa-meth 和 BitMapperBS)上进行测试后,我们发现总体上各检测方法之间具有高度一致性,但在读取映射、CpG 捕获、覆盖度和平台性能方面也存在差异,并且在 26 种微阵列归一化算法中性能不同。
本文提供的数据可以指导在表观遗传学研究中使用这些 DNA 参考材料,以及为未来研究中的实验设计提供最佳实践。通过利用七个被指定为公开可用参考材料的人类细胞系,这些数据可以用作推进表观遗传学研究的基线。