Billingsley Kimberley J, Meredith Melissa, Daida Kensuke, Jerez Pilar Alvarez, Negi Shloka, Malik Laksh, Genner Rylee M, Moller Abraham, Zheng Xinchang, Gibson Sophia B, Mastoras Mira, Baker Breeana, Kouam Cedric, Paquette Kimberly, Jarreau Paige, Makarious Mary B, Moore Anni, Hong Samantha, Vitale Dan, Shah Syed, Monlong Jean, Pantazis Caroline B, Asri Mobin, Shafin Kishwar, Carnevali Paolo, Marenco Stefano, Auluck Pavan, Mandal Ajeet, Miga Karen H, Rhie Arang, Reed Xylena, Ding Jinhui, Cookson Mark R, Nalls Mike, Singleton Andrew, Miller Danny E, Chaisson Mark, Timp Winston, Gibbs J Raphael, Phillippy Adam M, Kolmogorov Mikhail, Jain Miten, Sedlazeck Fritz J, Paten Benedict, Blauwendraat Cornelis
Center for Alzheimer's and Related Dementias, National Institute on Aging and National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA.
UC Santa Cruz Genomics Institute, Santa Cruz, CA, USA.
bioRxiv. 2024 Dec 17:2024.12.16.628723. doi: 10.1101/2024.12.16.628723.
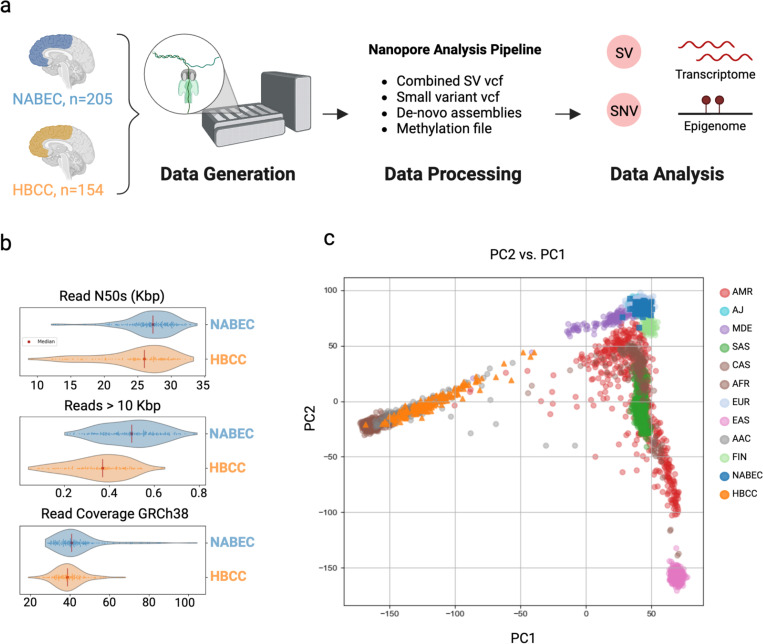
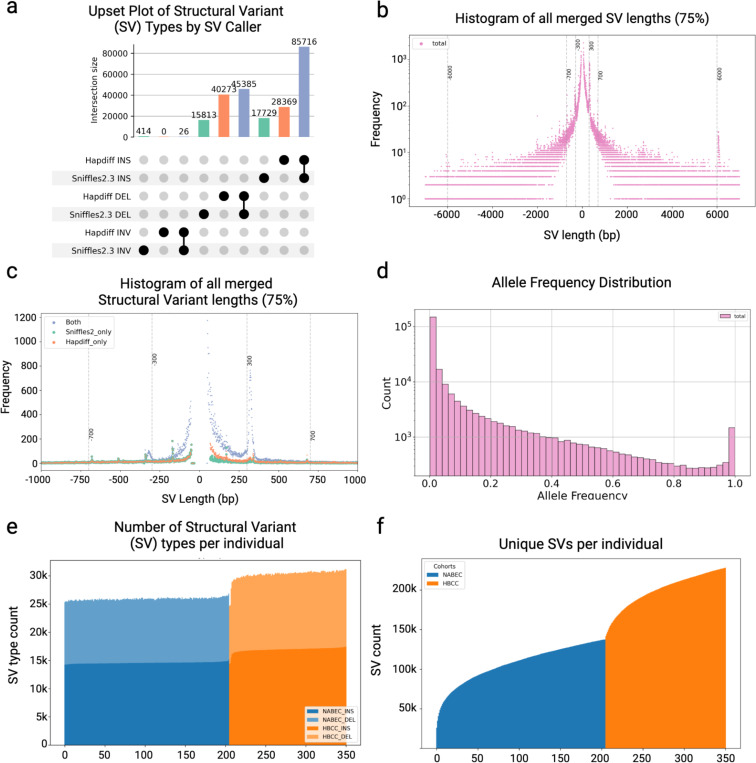
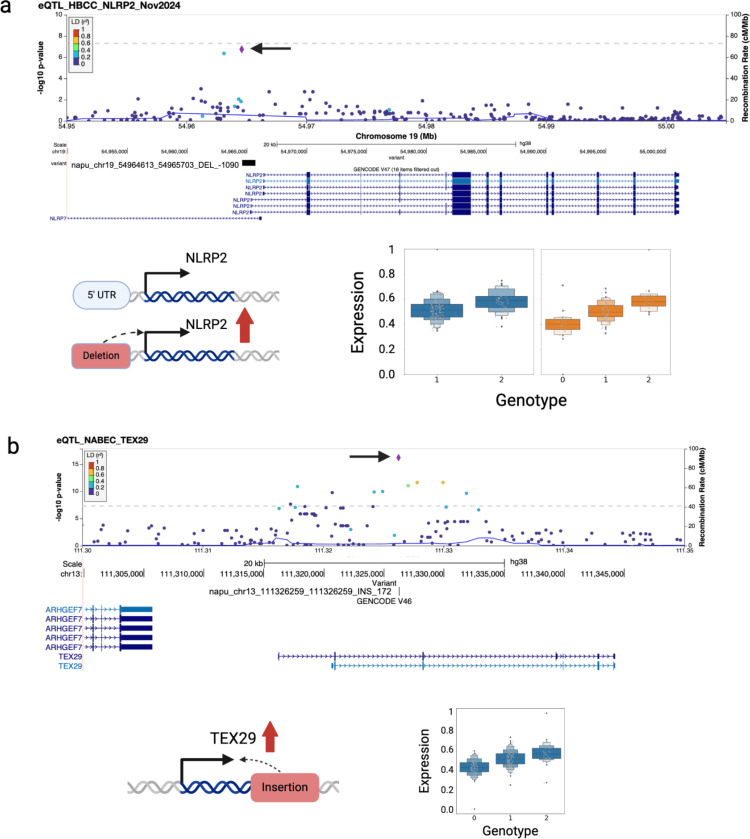
Structural variants (SVs) drive gene expression in the human brain and are causative of many neurological conditions. However, most existing genetic studies have been based on short-read sequencing methods, which capture fewer than half of the SVs present in any one individual. Long-read sequencing (LRS) enhances our ability to detect disease-associated and functionally relevant structural variants (SVs); however, its application in large-scale genomic studies has been limited by challenges in sample preparation and high costs. Here, we leverage a new scalable wet-lab protocol and computational pipeline for whole-genome Oxford Nanopore Technologies sequencing and apply it to neurologically normal control samples from the North American Brain Expression Consortium (NABEC) (European ancestry) and Human Brain Collection Core (HBCC) (African or African admixed ancestry) cohorts. Through this work, we present a publicly available long-read resource from 351 human brain samples (median N50: 27 Kbp and at an average depth of ~40x genome coverage). We discover approximately 234,905 SVs and produce locally phased assemblies that cover 95% of all protein-coding genes in GRCh38. Utilizing matched expression datasets for these samples, we apply quantitative trait locus (QTL) analyses and identify SVs that impact gene expression in post-mortem frontal cortex brain tissue. Further, we determine haplotype-specific methylation signatures at millions of CpGs and, with this data, identify cis-acting SVs. In summary, these results highlight that large-scale LRS can identify complex regulatory mechanisms in the brain that were inaccessible using previous approaches. We believe this new resource provides a critical step toward understanding the biological effects of genetic variation in the human brain.
结构变异(SVs)驱动人类大脑中的基因表达,并导致许多神经系统疾病。然而,大多数现有的基因研究都基于短读长测序方法,这种方法捕获的任何一个个体中存在的SVs不到一半。长读长测序(LRS)增强了我们检测与疾病相关和功能相关的结构变异(SVs)的能力;然而,其在大规模基因组研究中的应用受到样本制备挑战和高成本的限制。在这里,我们利用一种新的可扩展湿实验室方案和计算流程进行全基因组牛津纳米孔技术测序,并将其应用于来自北美大脑表达联盟(NABEC)(欧洲血统)和人类大脑收集核心(HBCC)(非洲或非洲混合血统)队列的神经正常对照样本。通过这项工作,我们展示了一个来自351个人类大脑样本的公开可用长读长资源(中位数N50:27 Kbp,平均基因组覆盖深度约为40倍)。我们发现了大约234,905个SVs,并生成了覆盖GRCh38中所有蛋白质编码基因95%的局部定相组装。利用这些样本的匹配表达数据集,我们进行数量性状基因座(QTL)分析,并识别影响死后额叶皮质脑组织中基因表达的SVs。此外,我们确定了数百万个CpG位点的单倍型特异性甲基化特征,并利用这些数据识别顺式作用SVs。总之,这些结果突出表明,大规模LRS可以识别以前方法无法触及的大脑中复杂的调控机制。我们相信,这个新资源为理解人类大脑中遗传变异的生物学效应迈出了关键一步。