Department of Infectious Diseases, Central Clinical School, Monash University, Melbourne, Victoria, Australia.
Department of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, The University of Melbourne, 792 Elizabeth Street, Melbourne, Victoria, Australia.
Microb Genom. 2021 Dec;7(12). doi: 10.1099/mgen.0.000694.
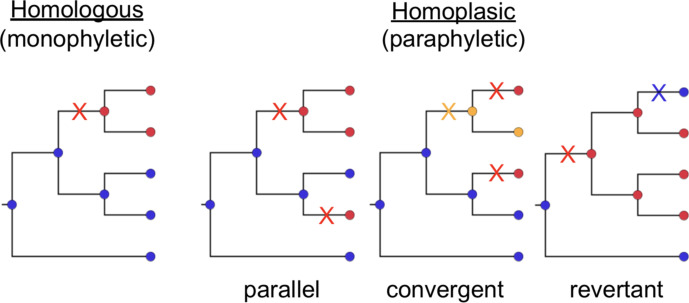
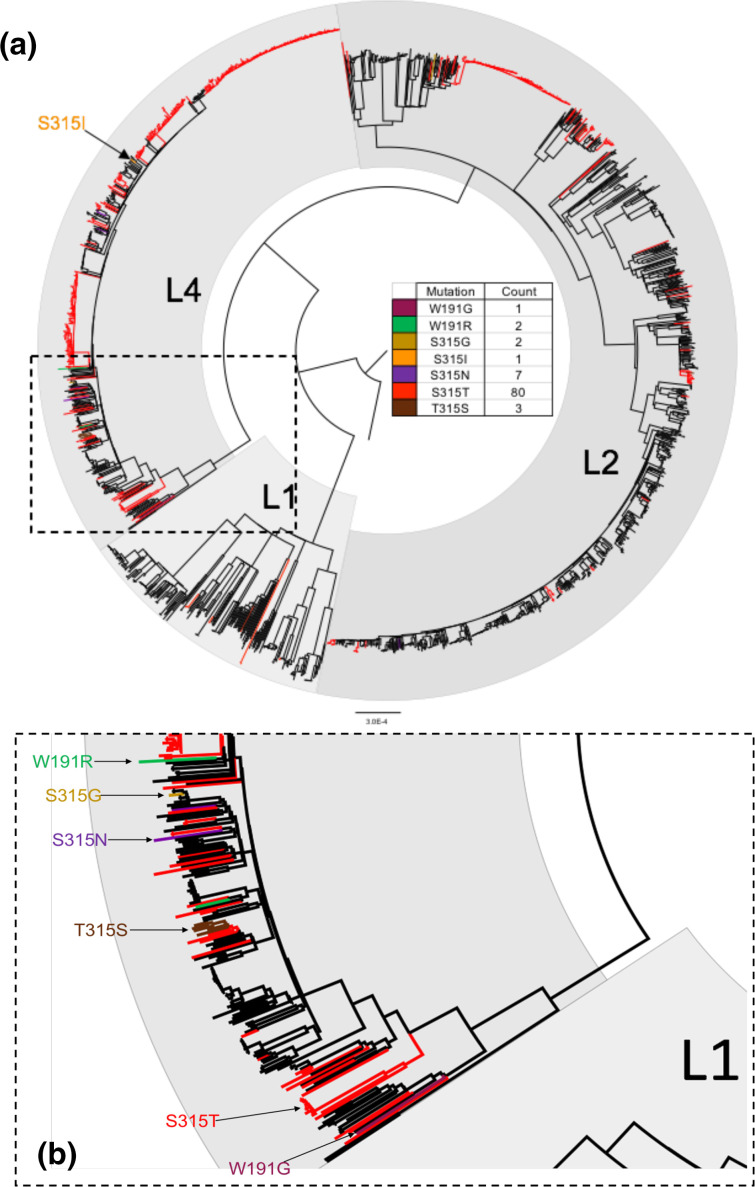
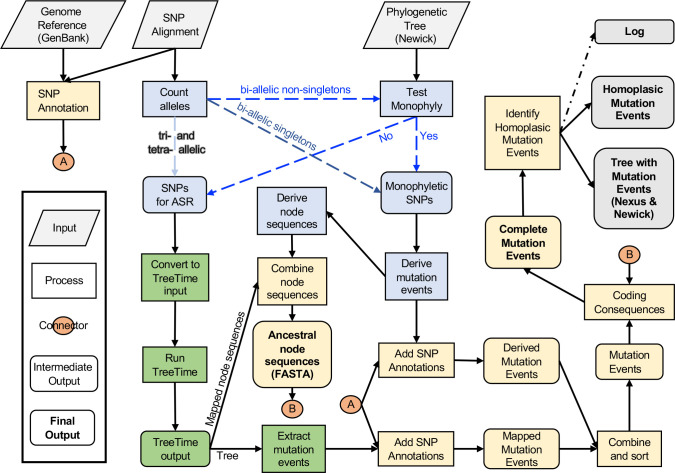
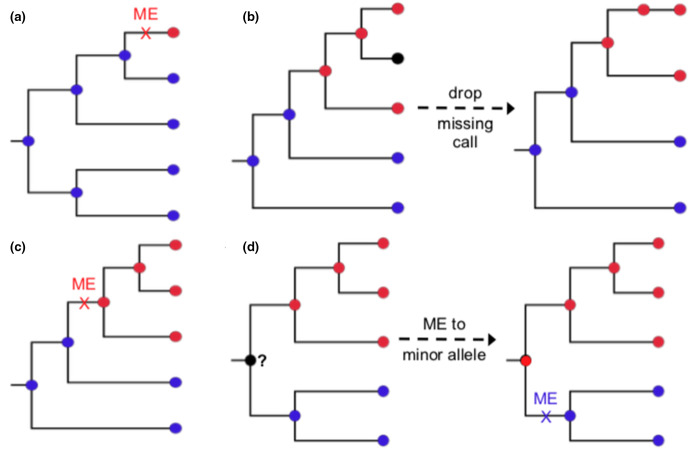
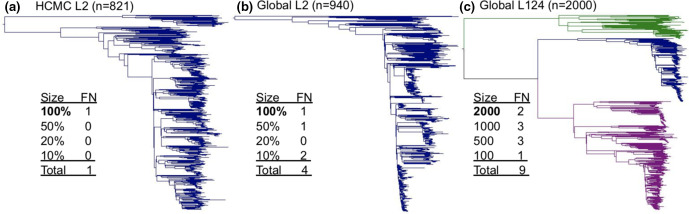
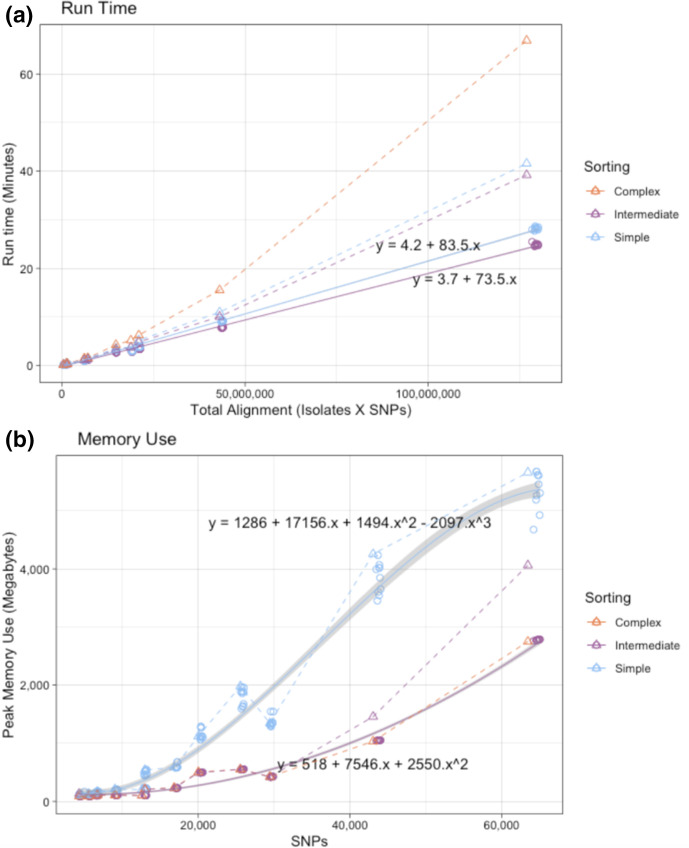
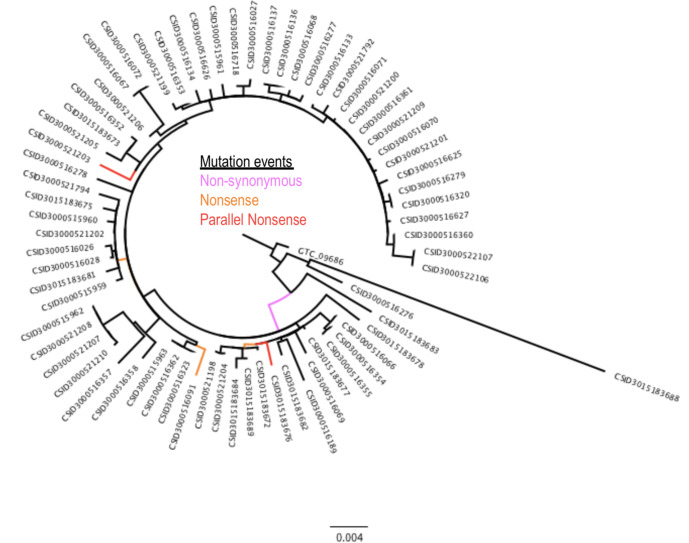
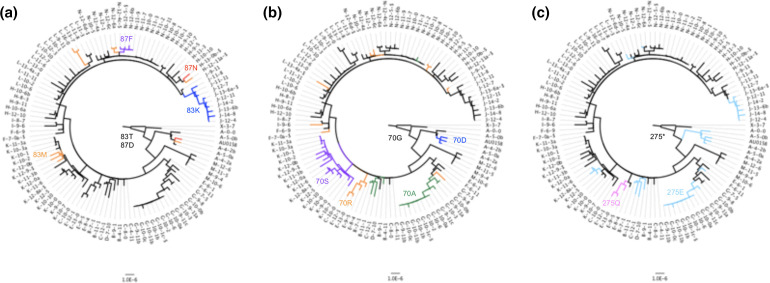
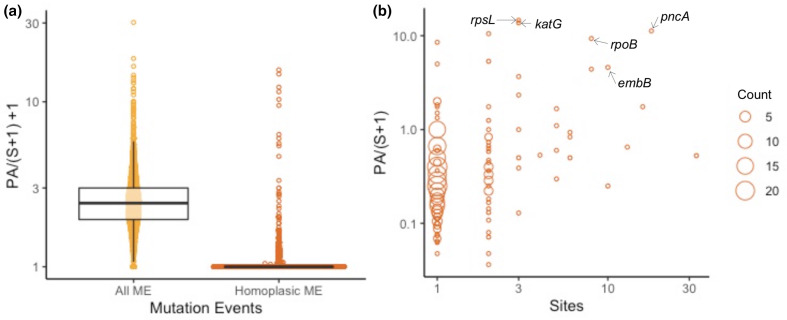
Homoplasic SNPs are considered important signatures of strong (positive) selective pressure, and hence of adaptive evolution for clinically relevant traits such as antibiotic resistance and virulence. Here we present a new tool, SNPPar, for efficient detection and analysis of homoplasic SNPs from large whole genome sequencing datasets (>1000 isolates and/or >100 000 SNPs). SNPPar takes as input an SNP alignment, tree and annotated reference genome, and uses a combination of simple monophyly tests and ancestral state reconstruction (ASR, via TreeTime) to assign mutation events to branches and identify homoplasies. Mutations are annotated at the level of codon and gene, to facilitate analysis of convergent evolution. Testing on simulated data (120 alignments representing local and global samples) showed SNPPar can detect homoplasic SNPs with very high specificity (zero false-positives in all tests) and high sensitivity (zero false-negatives in 89 % of tests). SNPPar analysis of three empirically sampled datasets (, and ) produced results that were in concordance with previous studies, in terms of both individual homoplasies and evidence of convergence at the codon and gene levels. SNPPar analysis of a simulated alignment of ~64 000 genome-wide SNPs from 2000 genomes took ~23 min and ~2.6 GB of RAM to generate complete annotated results on a laptop. This analysis required ASR be conducted for only 1.25 % of SNPs, and the ASR step took ~23 s and 0.4 GB of RAM. SNPPar automates the detection and annotation of homoplasic SNPs efficiently and accurately from large SNP alignments. As demonstrated by the examples included here, this information can be readily used to explore the role of homoplasy in parallel and/or convergent evolution at the level of nucleotide, codon and/or gene.
同态 SNP 被认为是强(正)选择压力的重要特征,因此也是与临床相关特征(如抗生素耐药性和毒力)相关的适应性进化的重要特征。在这里,我们提出了一种新的工具 SNPPar,用于从大型全基因组测序数据集(>1000 个分离株和/或>100000 个 SNPs)中高效检测和分析同态 SNP。SNPPar 以 SNP 比对、树和注释的参考基因组作为输入,使用简单的单系测试和祖先状态重建(通过 TreeTime)的组合,将突变事件分配给分支并识别同态性。突变在密码子和基因水平上进行注释,以方便对趋同进化进行分析。在模拟数据(代表局部和全局样本的 120 个比对)上的测试表明,SNPPar 可以以非常高的特异性(所有测试均无假阳性)和高灵敏度(89%的测试均无假阴性)检测同态 SNP。对三个经验采样数据集(1000 个细菌基因组、1000 个结核分枝杆菌基因组和 33 个淋病奈瑟氏球菌基因组)的 SNPPar 分析结果与以前的研究一致,无论是在个体同态性方面,还是在密码子和基因水平上的趋同证据方面。对来自 2000 个基因组的约 64000 个全基因组 SNP 的模拟比对进行的 SNPPar 分析,在笔记本电脑上生成完整注释结果需要约 23 分钟和 2.6GB 的 RAM。此分析仅需要对 1.25%的 SNPs 进行 ASR,ASR 步骤需要约 23 秒和 0.4GB 的 RAM。SNPPar 可以有效地从大型 SNP 比对中自动检测和注释同态 SNP,并且准确无误。正如这里包含的示例所示,这些信息可以很容易地用于探索同态性在核苷酸、密码子和/或基因水平上的平行和/或趋同进化中的作用。