Biomedical Informatics Training Program, Stanford University, Stanford, CA 94305, USA.
Pac Symp Biocomput. 2022;27:10-21.
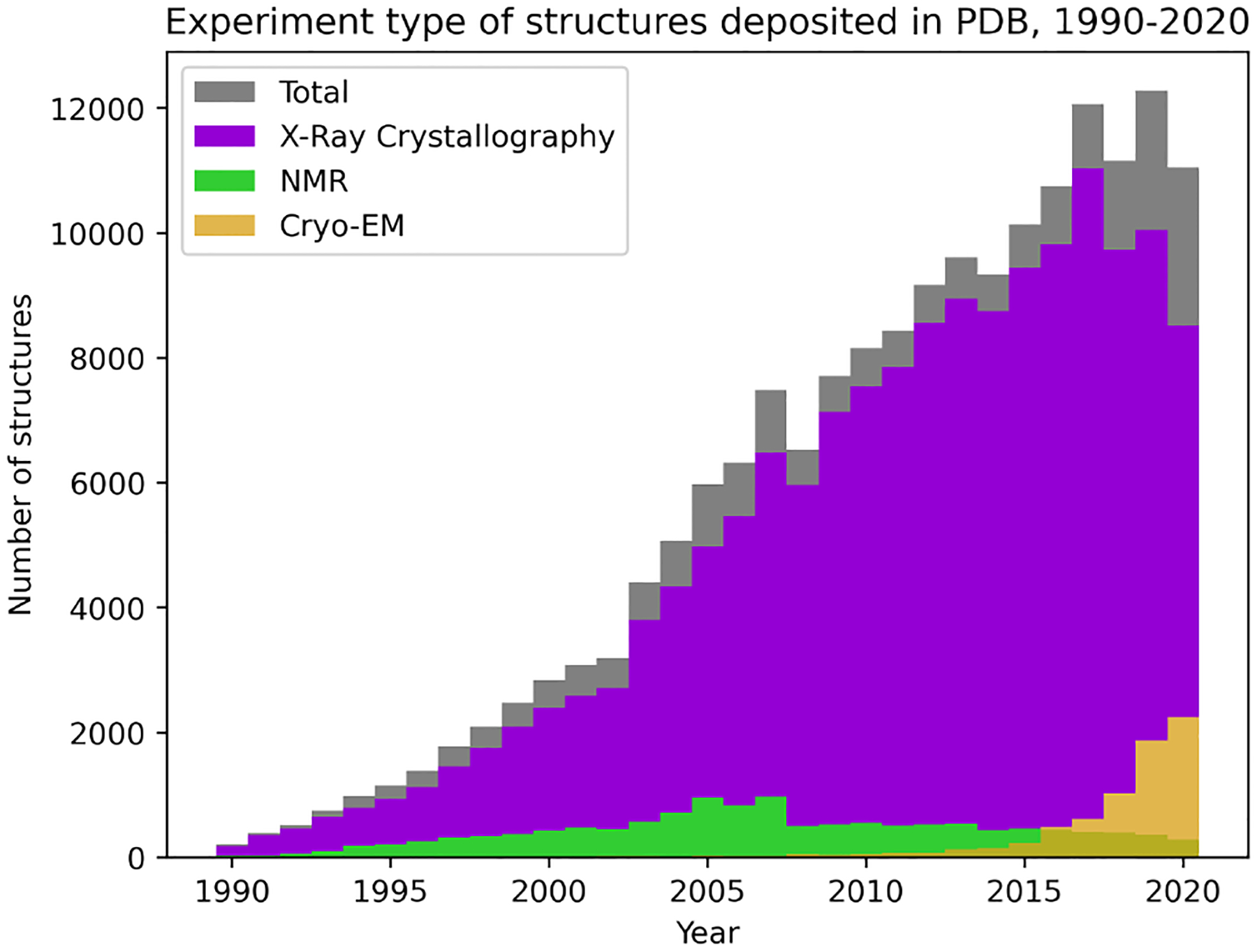
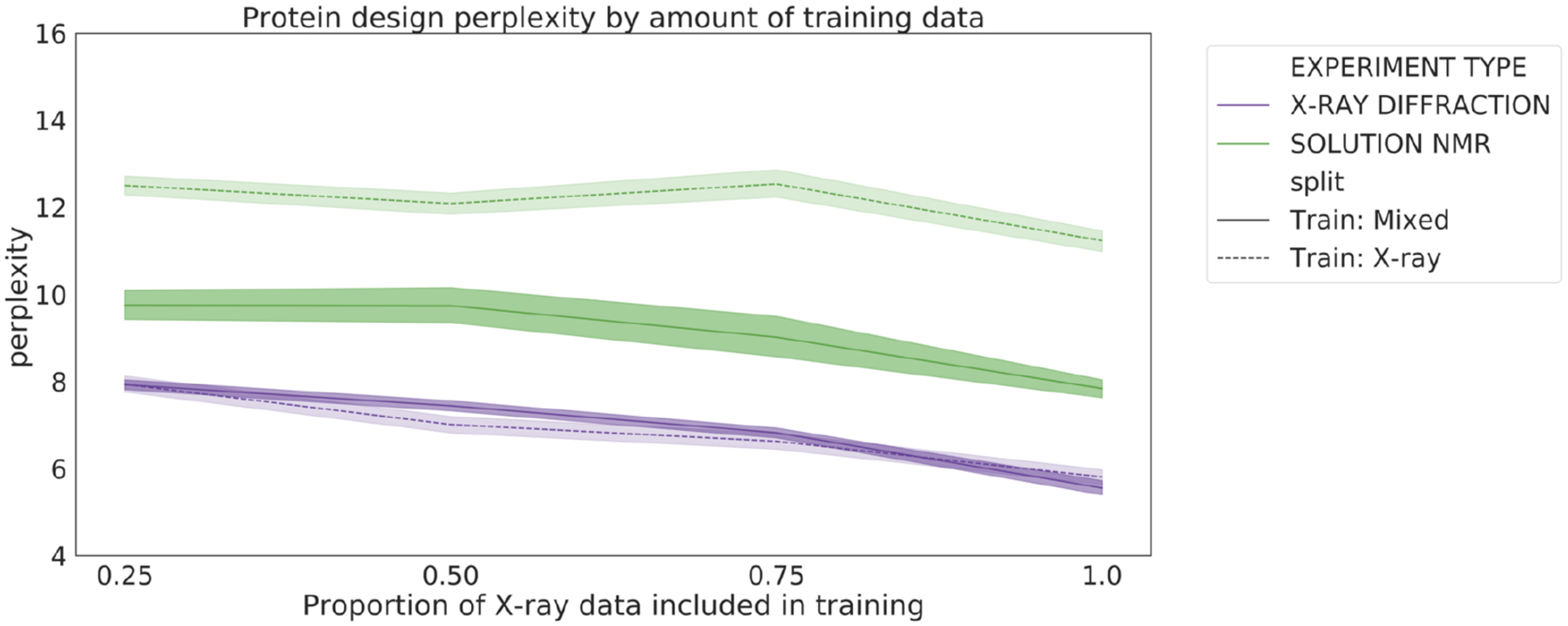
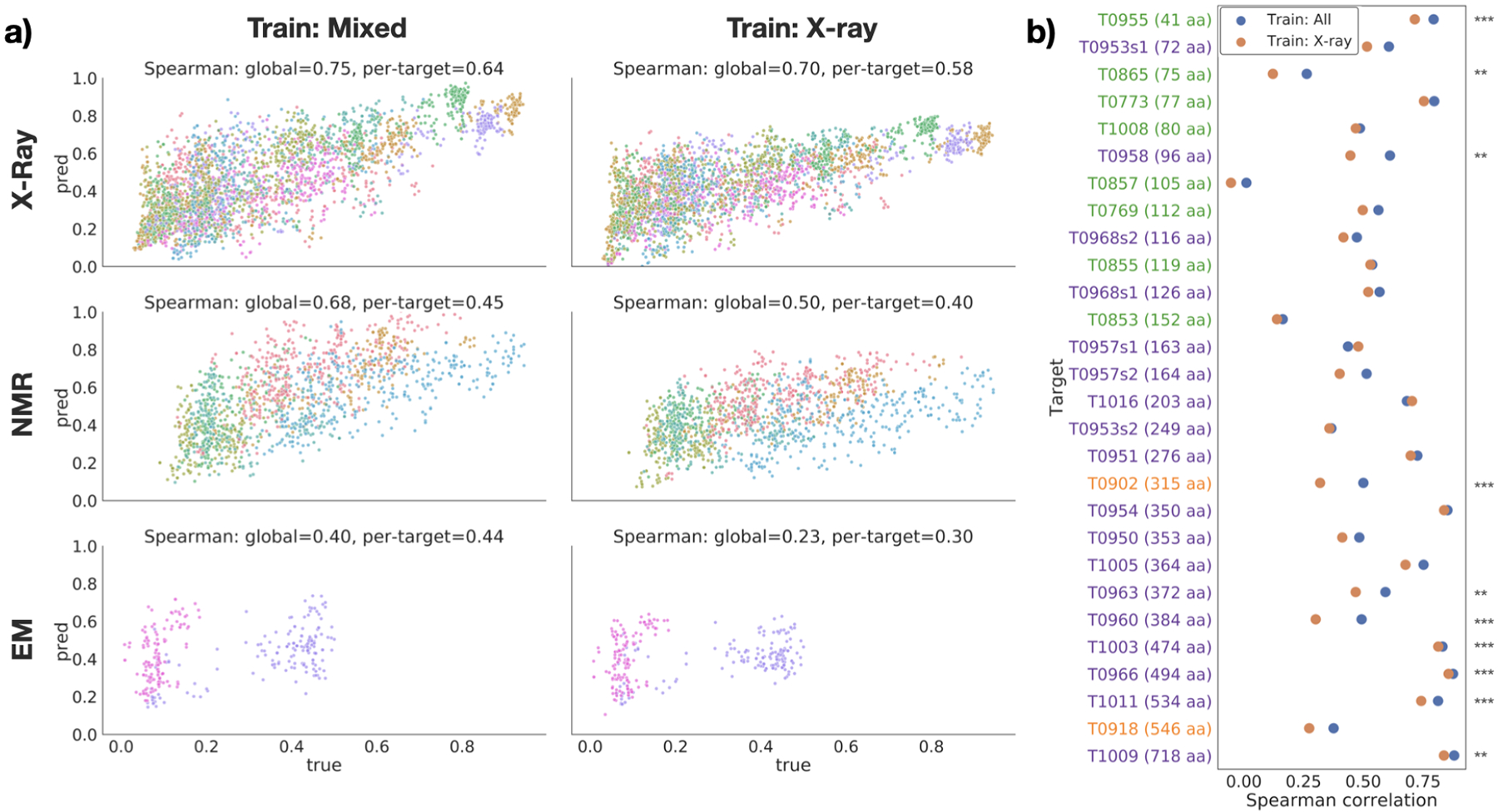
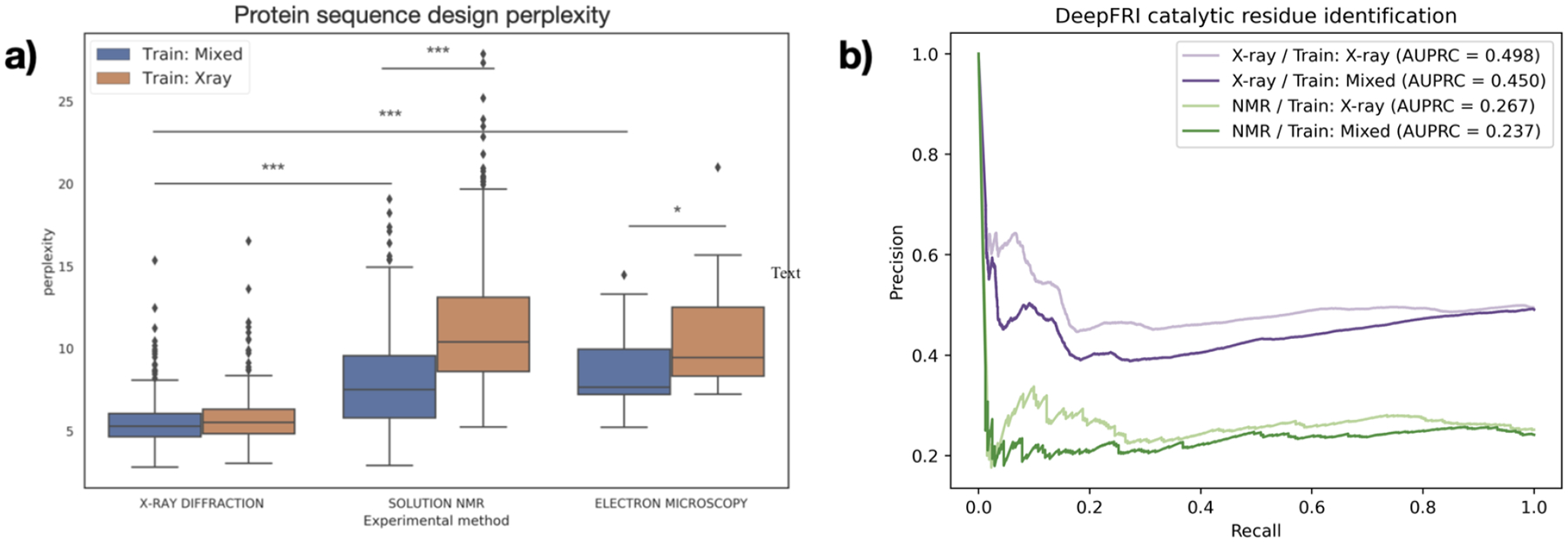
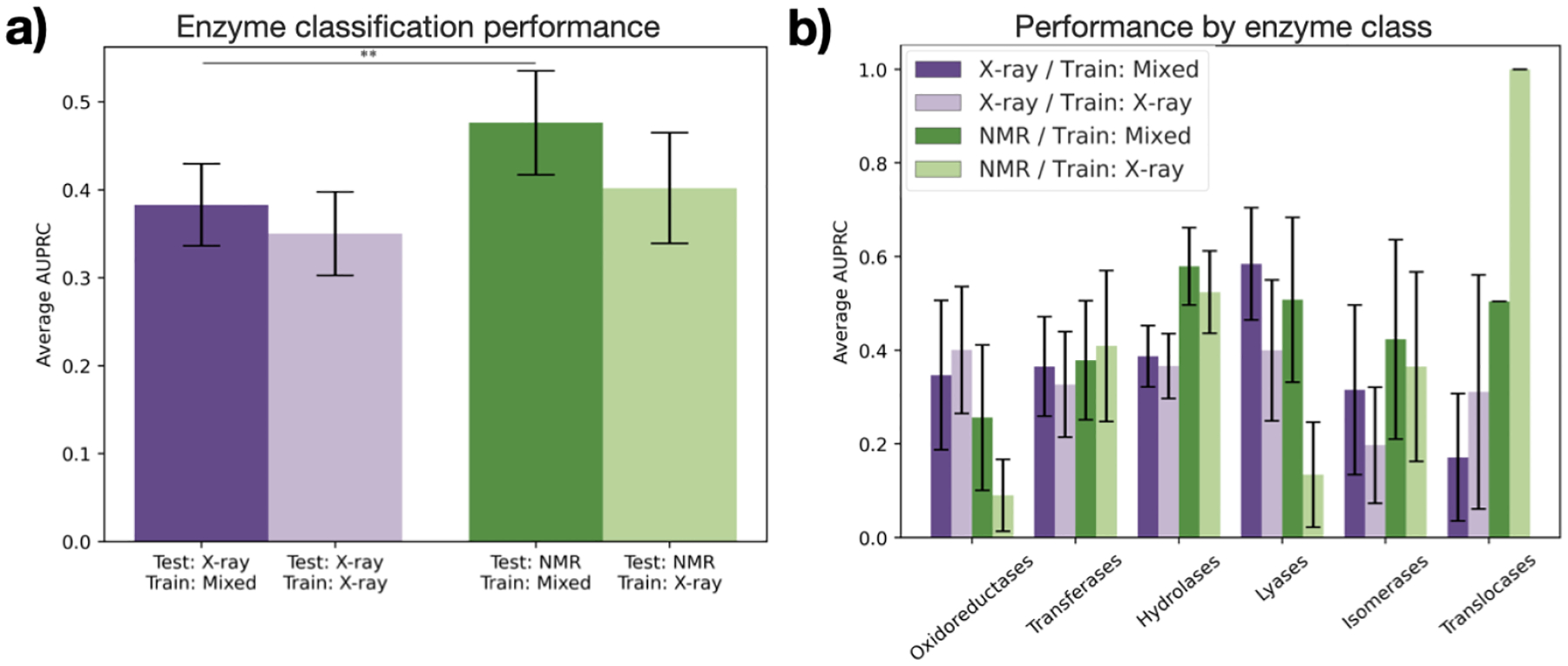
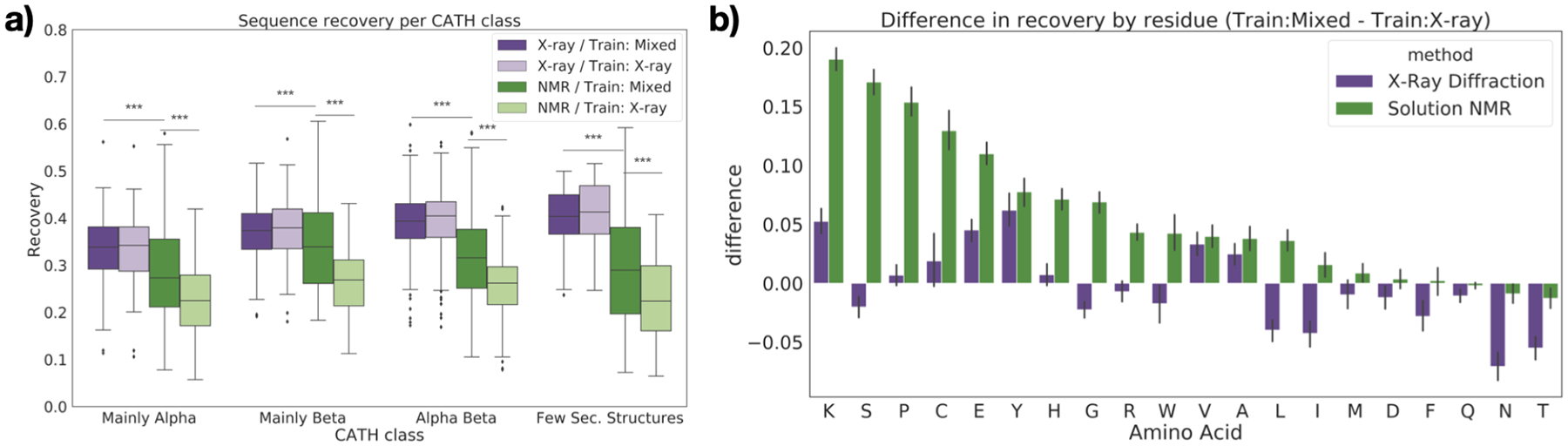
The three-dimensional structures of proteins are crucial for understanding their molecular mechanisms and interactions. Machine learning algorithms that are able to learn accurate representations of protein structures are therefore poised to play a key role in protein engineering and drug development. The accuracy of such models in deployment is directly influenced by training data quality. The use of different experimental methods for protein structure determination may introduce bias into the training data. In this work, we evaluate the magnitude of this effect across three distinct tasks: estimation of model accuracy, protein sequence design, and catalytic residue prediction. Most protein structures are derived from X-ray crystallography, nuclear magnetic resonance (NMR), or cryo-electron microscopy (cryo-EM); we trained each model on datasets consisting of either all three structure types or of only X-ray data. We Find that across these tasks, models consistently perform worse on test sets derived from NMR and cryo-EM than they do on test sets of structures derived from X-ray crystallography, but that the difference can be mitigated when NMR and cryo-EM structures are included in the training set. Importantly, we show that including all three types of structures in the training set does not degrade test performance on X-ray structures, and in some cases even increases it. Finally, we examine the relationship between model performance and the biophysical properties of each method, and recommend that the biochemistry of the task of interest should be considered when composing training sets.
蛋白质的三维结构对于理解其分子机制和相互作用至关重要。因此,能够学习蛋白质结构准确表示的机器学习算法有望在蛋白质工程和药物开发中发挥关键作用。这些模型在部署中的准确性直接受到训练数据质量的影响。不同的实验方法用于蛋白质结构测定可能会给训练数据带来偏差。在这项工作中,我们在三个不同的任务中评估了这种影响的大小:模型准确性估计、蛋白质序列设计和催化残基预测。大多数蛋白质结构来自 X 射线晶体学、核磁共振(NMR)或低温电子显微镜(cryo-EM);我们在数据集上训练了每个模型,这些数据集由所有三种结构类型或仅 X 射线数据组成。我们发现,在这些任务中,模型在来自 NMR 和 cryo-EM 的测试集上的表现始终不如来自 X 射线晶体学的测试集上的表现差,但当 NMR 和 cryo-EM 结构包含在训练集中时,这种差异可以减轻。重要的是,我们表明在训练集中包含所有三种类型的结构不会降低 X 射线结构的测试性能,在某些情况下甚至会提高它。最后,我们研究了模型性能与每种方法的生物物理特性之间的关系,并建议在组成训练集时应考虑感兴趣任务的生物化学特性。