NCI-Designated Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, USA.
Conrad Prebys Center for Chemical Genomics, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, USA.
J Biol Chem. 2022 Jan;298(1):101477. doi: 10.1016/j.jbc.2021.101477. Epub 2021 Dec 10.
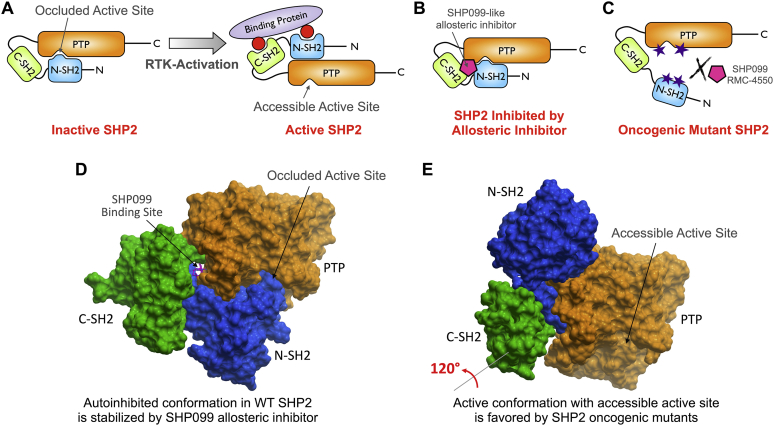
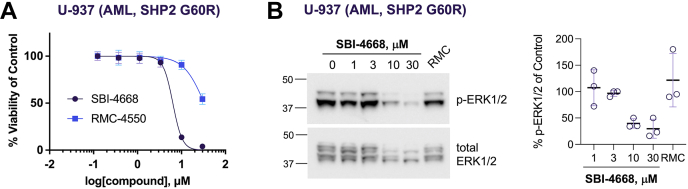
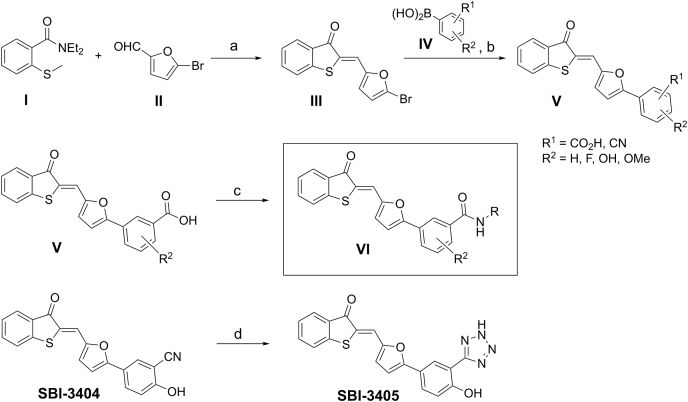
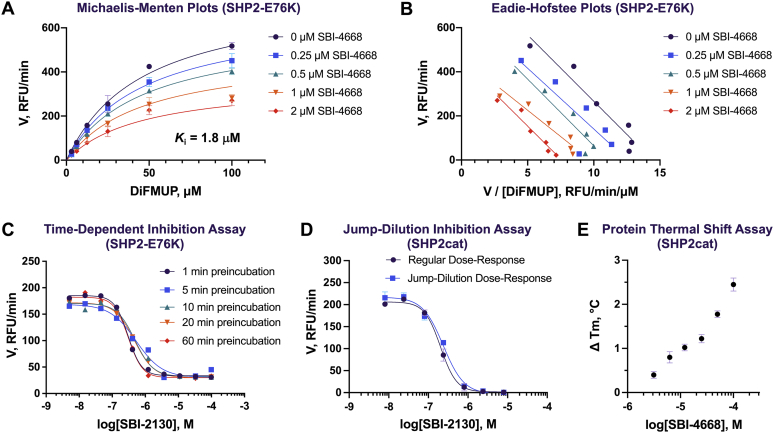
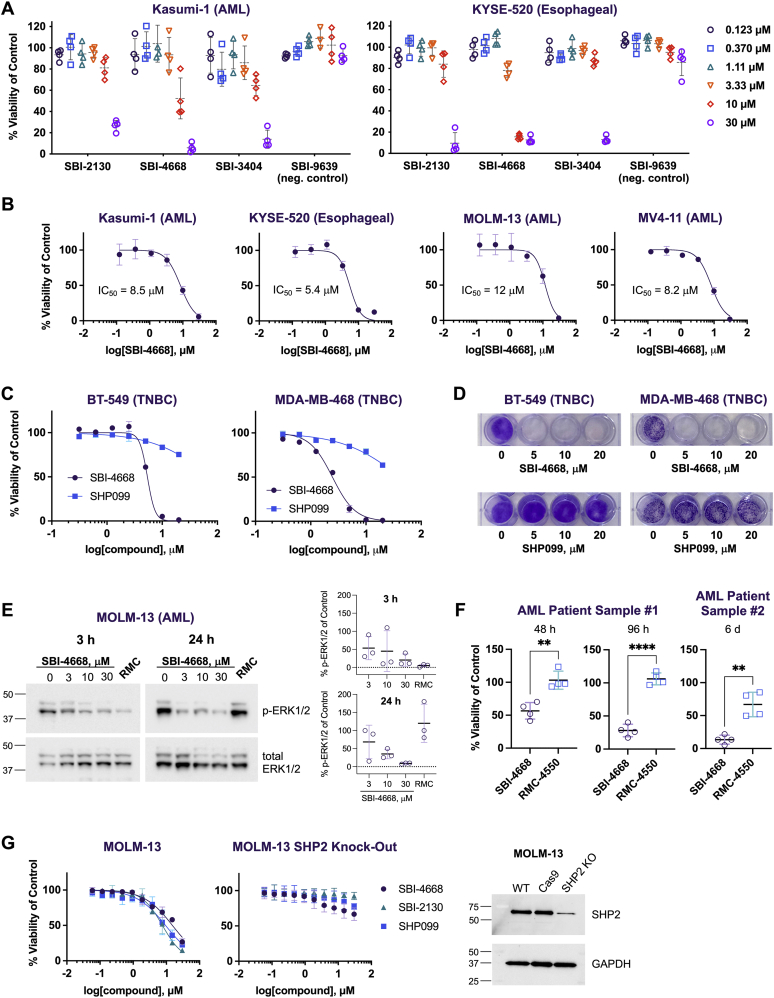
Disturbance of the dynamic balance between tyrosine phosphorylation and dephosphorylation of signaling molecules, controlled by protein tyrosine kinases and protein tyrosine phosphatases (PTPs), is known to lead to the development of cancer. While most approved targeted cancer therapies are tyrosine kinase inhibitors, PTPs have long been stigmatized as undruggable and have only recently gained renewed attention in drug discovery. One PTP target is the Src-homology 2 domain-containing phosphatase 2 (SHP2). SHP2 is implicated in tumor initiation, progression, metastasis, and treatment resistance, primarily because of its role as a signaling nexus of the extracellular signal-regulated kinase pathway, acting upstream of the small GTPase Ras. Efforts to develop small molecules that target SHP2 are ongoing, and several SHP2 allosteric inhibitors are currently in clinical trials for the treatment of solid tumors. However, while the reported allosteric inhibitors are highly effective against cells expressing WT SHP2, none have significant activity against the most frequent oncogenic SHP2 variants that drive leukemogenesis in several juvenile and acute leukemias. Here, we report the discovery of novel furanylbenzamide molecules as inhibitors of both WT and oncogenic SHP2. Importantly, these inhibitors readily cross cell membranes, bind and inhibit SHP2 under physiological conditions, and effectively decrease the growth of cancer cells, including triple-negative breast cancer cells, acute myeloid leukemia cells expressing either WT or oncogenic SHP2, and patient-derived acute myeloid leukemia cells. These novel compounds are effective chemical probes of active SHP2 and may serve as starting points for therapeutics targeting WT or mutant SHP2 in cancer.
信号分子的酪氨酸磷酸化和去磷酸化的动态平衡被蛋白酪氨酸激酶和蛋白酪氨酸磷酸酶(PTPs)所控制,其紊乱会导致癌症的发生。虽然大多数已批准的靶向癌症疗法是酪氨酸激酶抑制剂,但 PTP 长期以来一直被污名化为不可成药的靶点,直到最近才在药物发现中重新受到关注。PTP 的一个靶点是含Src 同源 2 结构域的磷酸酶 2(SHP2)。SHP2 参与肿瘤的起始、进展、转移和治疗耐药性,主要是因为它作为细胞外信号调节激酶途径的信号枢纽,作用于小 GTP 酶 Ras 的上游。开发靶向 SHP2 的小分子的努力正在进行中,目前有几种 SHP2 别构抑制剂正在进行治疗实体瘤的临床试验。然而,虽然报道的别构抑制剂对表达 WT SHP2 的细胞非常有效,但对驱动几种青少年和急性白血病中白血病发生的最常见致癌 SHP2 变体均没有显著活性。在这里,我们报告了新型呋喃苯甲酰胺分子作为 WT 和致癌 SHP2 的抑制剂的发现。重要的是,这些抑制剂很容易穿过细胞膜,在生理条件下结合并抑制 SHP2,并有效地抑制包括三阴性乳腺癌细胞、表达 WT 或致癌 SHP2 的急性髓系白血病细胞和患者来源的急性髓系白血病细胞在内的癌细胞的生长。这些新型化合物是活性 SHP2 的有效化学探针,可能成为针对癌症中 WT 或突变 SHP2 的治疗方法的起点。