Wang Jinan, Arantes Pablo R, Bhattarai Apurba, Hsu Rohaine V, Pawnikar Shristi, Huang Yu-Ming M, Palermo Giulia, Miao Yinglong
Center for Computational Biology and Department of Molecular Biosciences, University of Kansas, 2030 Becker Dr., Lawrence, KS, 66047, United States.
Department of Bioengineering, University of California Riverside, 900 University Avenue, Riverside, CA 92512, United States.
Wiley Interdiscip Rev Comput Mol Sci. 2021 Sep-Oct;11(5). doi: 10.1002/wcms.1521. Epub 2021 Mar 1.
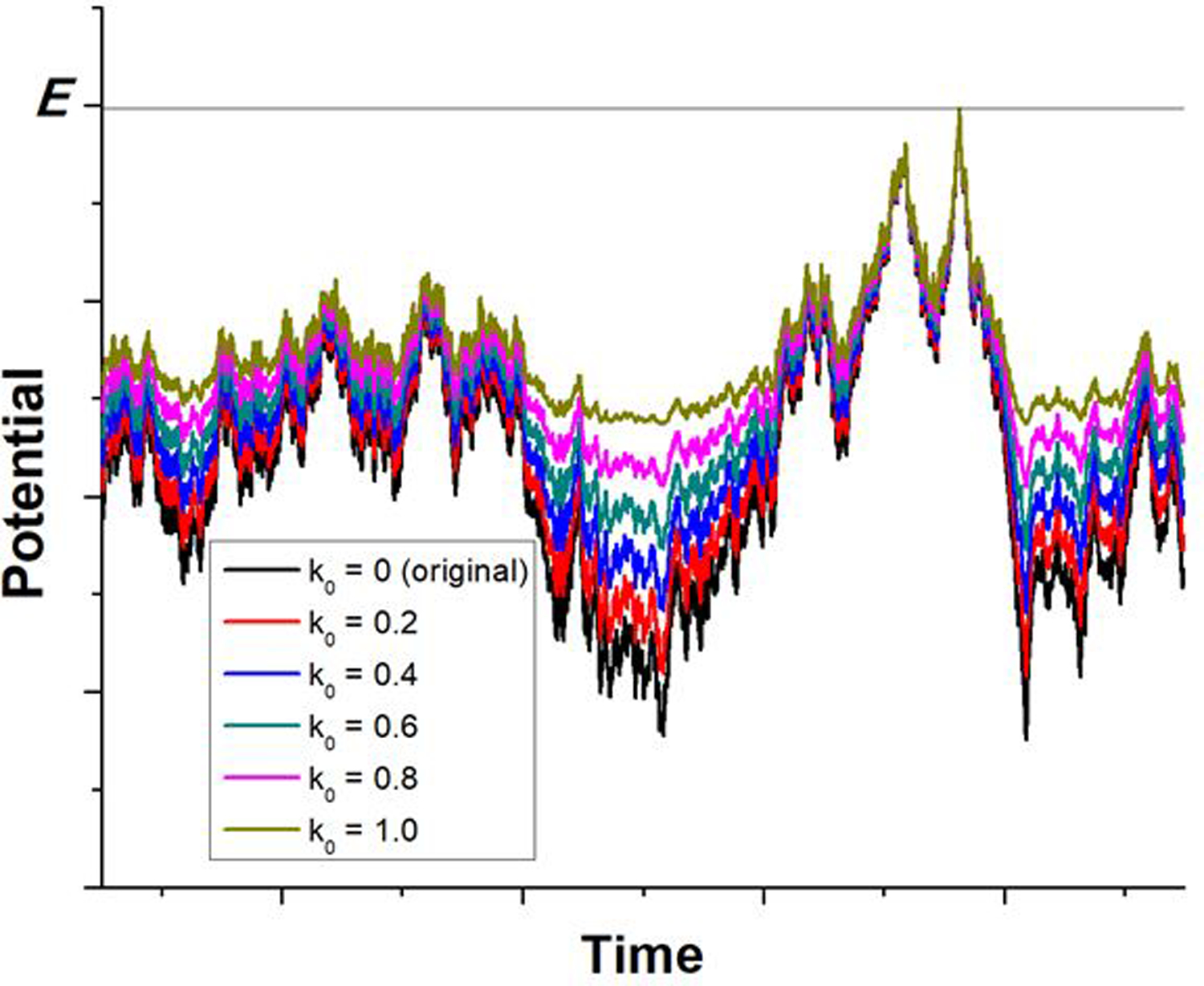
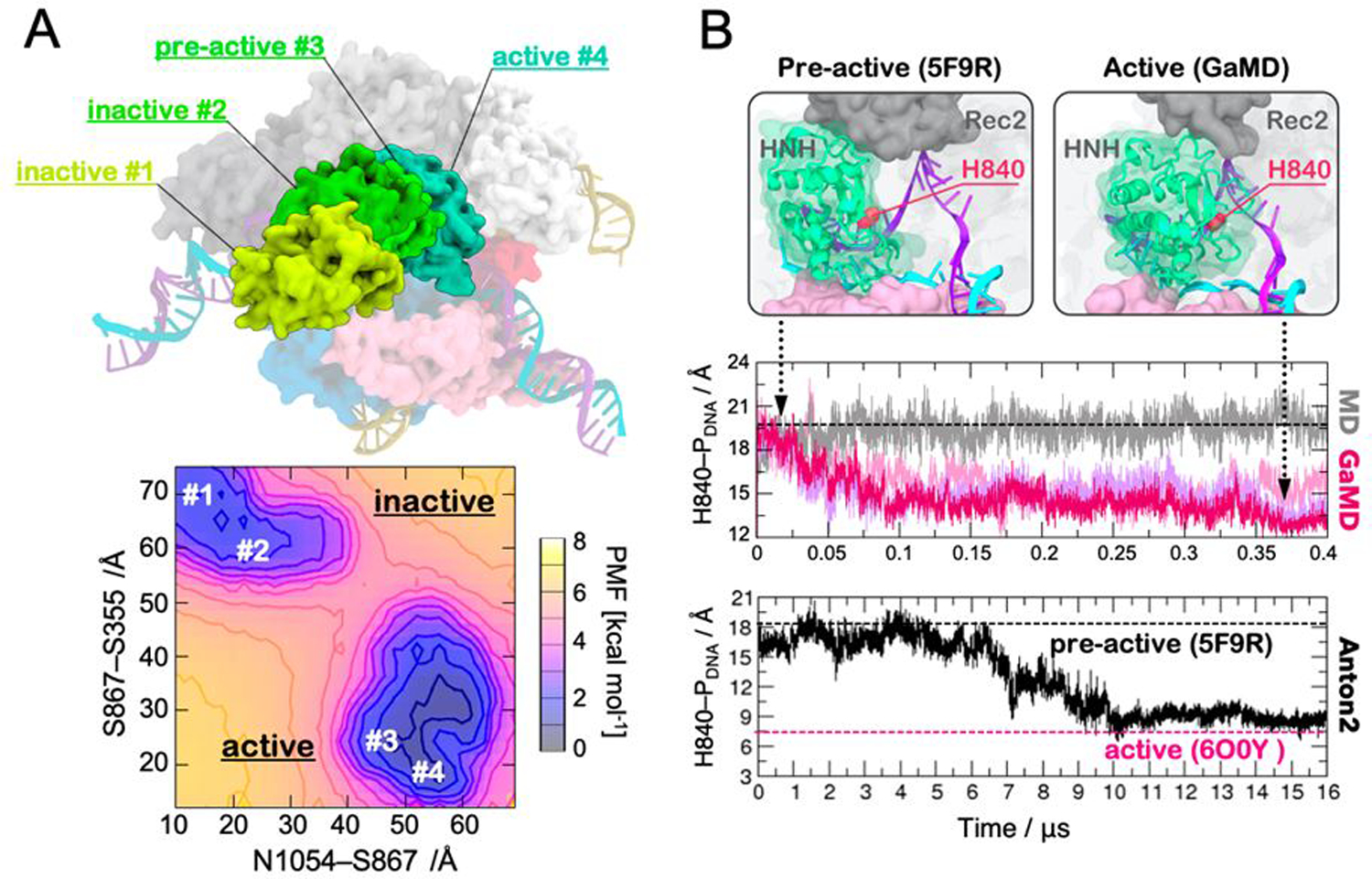
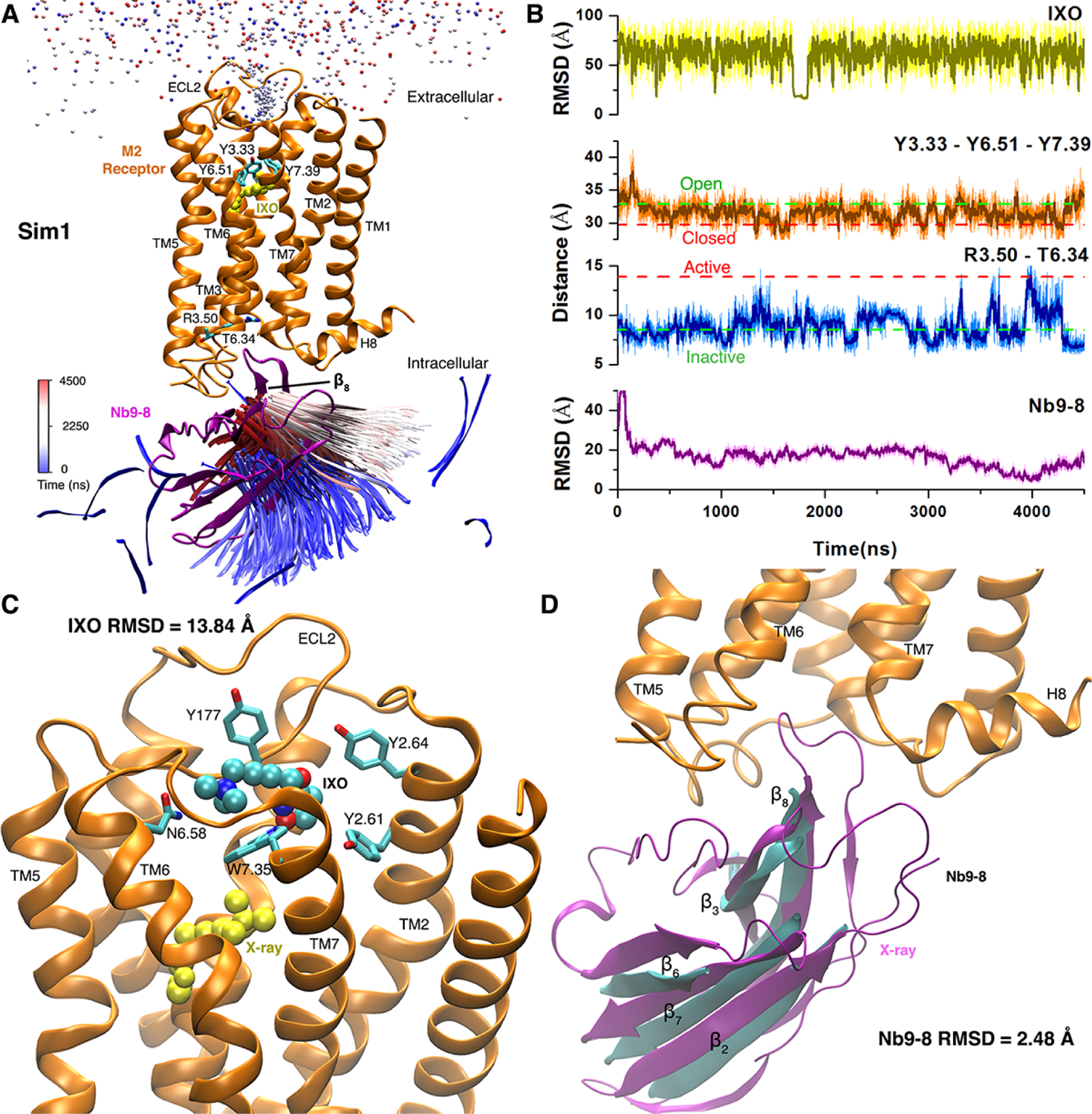
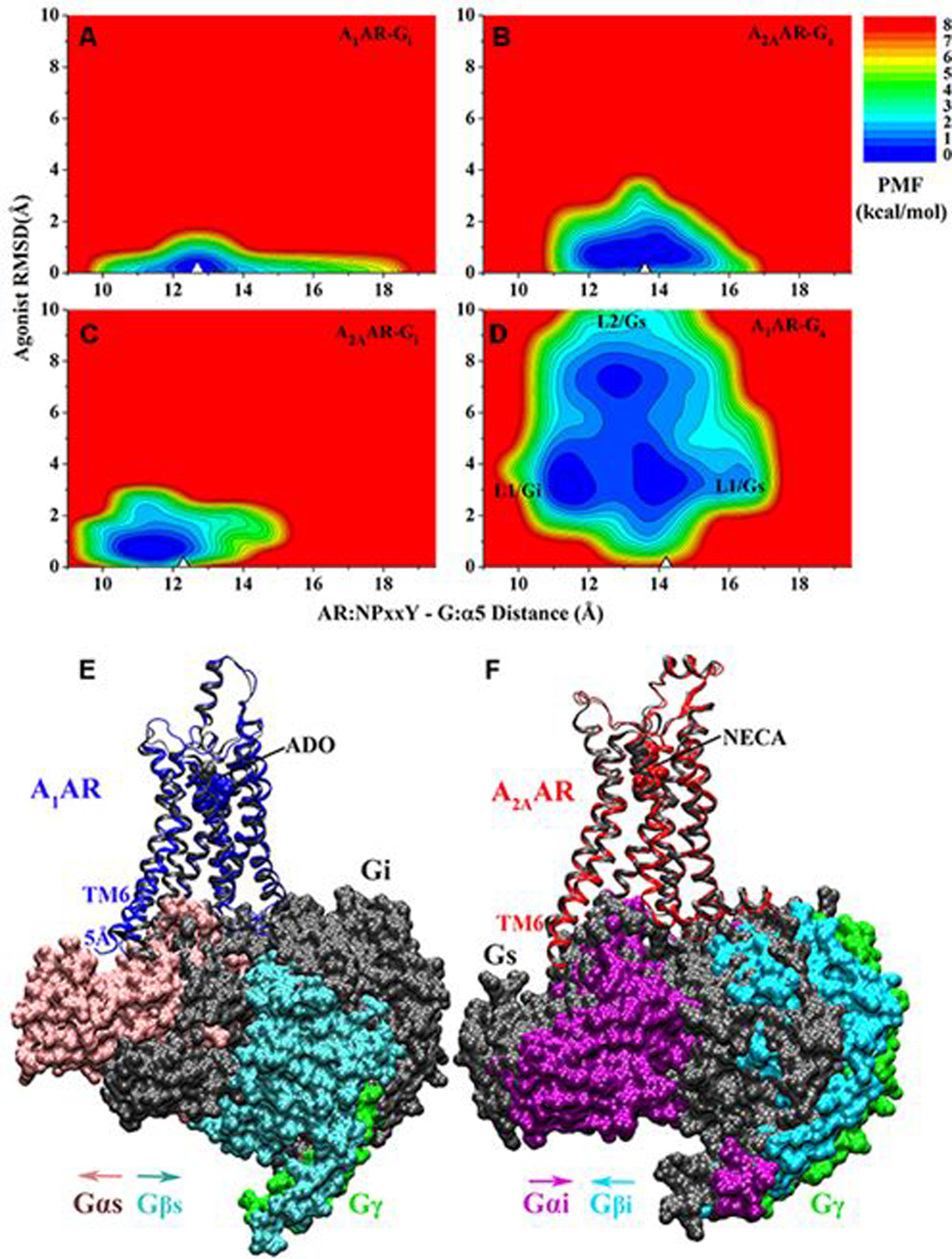
Gaussian accelerated molecular dynamics (GaMD) is a robust computational method for simultaneous unconstrained enhanced sampling and free energy calculations of biomolecules. It works by adding a harmonic boost potential to smooth biomolecular potential energy surface and reduce energy barriers. GaMD greatly accelerates biomolecular simulations by orders of magnitude. Without the need to set predefined reaction coordinates or collective variables, GaMD provides unconstrained enhanced sampling and is advantageous for simulating complex biological processes. The GaMD boost potential exhibits a Gaussian distribution, thereby allowing for energetic reweighting via cumulant expansion to the second order (i.e., "Gaussian approximation"). This leads to accurate reconstruction of free energy landscapes of biomolecules. Hybrid schemes with other enhanced sampling methods, such as the replica exchange GaMD (rex-GaMD) and replica exchange umbrella sampling GaMD (GaREUS), have also been introduced, further improving sampling and free energy calculations. Recently, new "selective GaMD" algorithms including the ligand GaMD (LiGaMD) and peptide GaMD (Pep-GaMD) enabled microsecond simulations to capture repetitive dissociation and binding of small-molecule ligands and highly flexible peptides. The simulations then allowed highly efficient quantitative characterization of the ligand/peptide binding thermodynamics and kinetics. Taken together, GaMD and its innovative variants are applicable to simulate a wide variety of biomolecular dynamics, including protein folding, conformational changes and allostery, ligand binding, peptide binding, protein-protein/nucleic acid/carbohydrate interactions, and carbohydrate/nucleic acid interactions. In this review, we present principles of the GaMD algorithms and recent applications in biomolecular simulations and drug design.
高斯加速分子动力学(GaMD)是一种强大的计算方法,用于对生物分子进行无约束增强采样和自由能计算。它通过添加一个谐波增强势来平滑生物分子势能面并降低能垒。GaMD能将生物分子模拟加速几个数量级。无需设置预定义的反应坐标或集体变量,GaMD提供无约束增强采样,有利于模拟复杂的生物过程。GaMD增强势呈现高斯分布,从而允许通过累积量展开到二阶(即“高斯近似”)进行能量重加权。这导致能够准确重建生物分子的自由能景观。还引入了与其他增强采样方法的混合方案,如副本交换GaMD(rex-GaMD)和副本交换伞形采样GaMD(GaREUS),进一步改进了采样和自由能计算。最近,包括配体GaMD(LiGaMD)和肽GaMD(Pep-GaMD)在内的新“选择性GaMD”算法实现了微秒级模拟,以捕捉小分子配体和高度柔性肽的重复解离和结合。这些模拟随后能够高效地对配体/肽结合的热力学和动力学进行定量表征。总之,GaMD及其创新变体适用于模拟各种生物分子动力学,包括蛋白质折叠、构象变化和变构、配体结合、肽结合、蛋白质-蛋白质/核酸/碳水化合物相互作用以及碳水化合物/核酸相互作用。在本综述中,我们介绍了GaMD算法的原理及其在生物分子模拟和药物设计中的最新应用。