Pang Yui Tik, Miao Yinglong, Wang Yi, McCammon J Andrew
Department of Physics, The Chinese University of Hong Kong , Shatin, New Territories, Hong Kong.
J Chem Theory Comput. 2017 Jan 10;13(1):9-19. doi: 10.1021/acs.jctc.6b00931. Epub 2016 Dec 30.
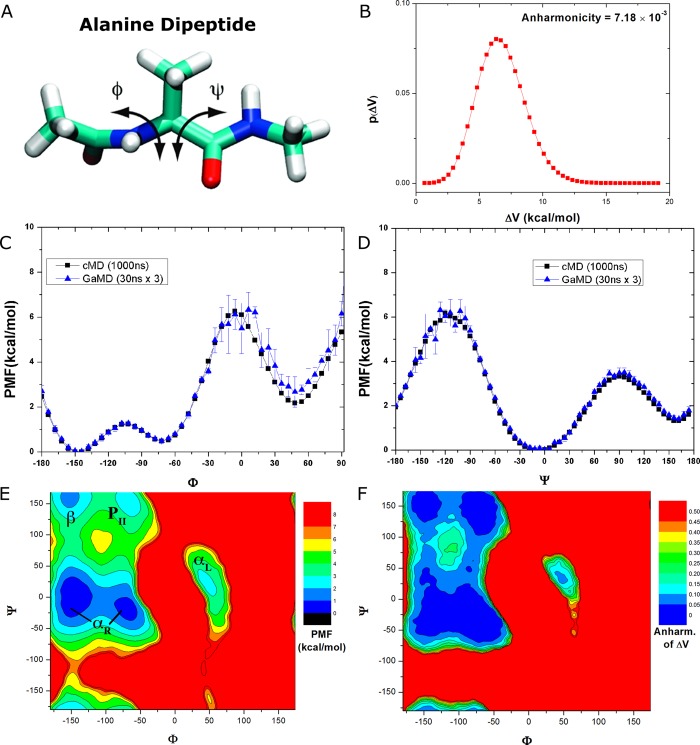
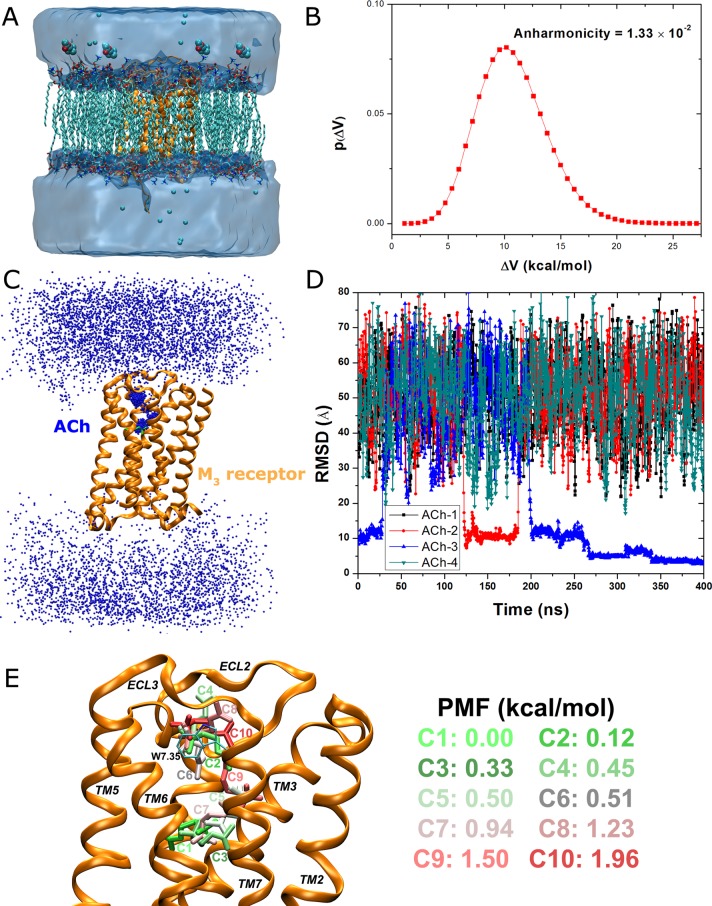
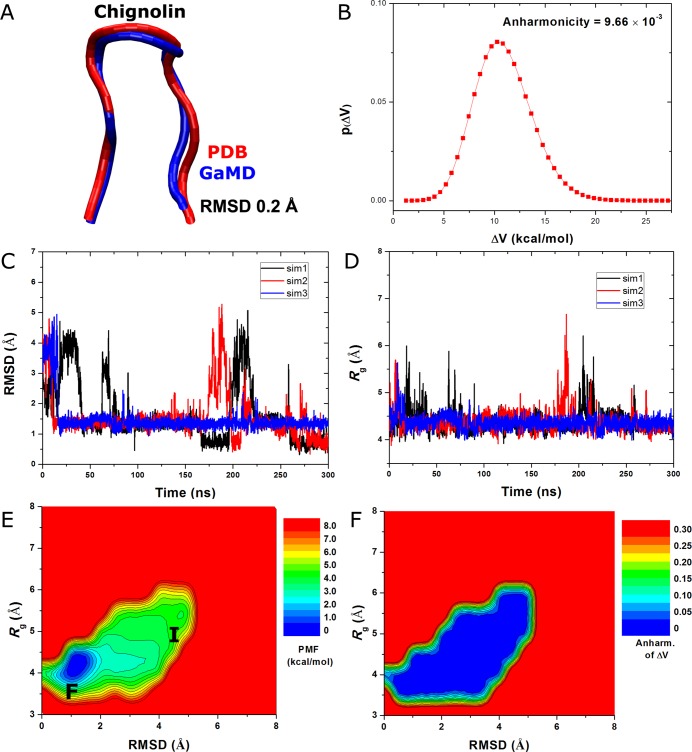
Gaussian accelerated molecular dynamics (GaMD) is a recently developed enhanced sampling technique that provides efficient free energy calculations of biomolecules. Like the previous accelerated molecular dynamics (aMD), GaMD allows for "unconstrained" enhanced sampling without the need to set predefined collective variables and so is useful for studying complex biomolecular conformational changes such as protein folding and ligand binding. Furthermore, because the boost potential is constructed using a harmonic function that follows Gaussian distribution in GaMD, cumulant expansion to the second order can be applied to recover the original free energy profiles of proteins and other large biomolecules, which solves a long-standing energetic reweighting problem of the previous aMD method. Taken together, GaMD offers major advantages for both unconstrained enhanced sampling and free energy calculations of large biomolecules. Here, we have implemented GaMD in the NAMD package on top of the existing aMD feature and validated it on three model systems: alanine dipeptide, the chignolin fast-folding protein, and the M muscarinic G protein-coupled receptor (GPCR). For alanine dipeptide, while conventional molecular dynamics (cMD) simulations performed for 30 ns are poorly converged, GaMD simulations of the same length yield free energy profiles that agree quantitatively with those of 1000 ns cMD simulation. Further GaMD simulations have captured folding of the chignolin and binding of the acetylcholine (ACh) endogenous agonist to the M muscarinic receptor. The reweighted free energy profiles are used to characterize the protein folding and ligand binding pathways quantitatively. GaMD implemented in the scalable NAMD is widely applicable to enhanced sampling and free energy calculations of large biomolecules.
高斯加速分子动力学(GaMD)是一种最近开发的增强采样技术,可对生物分子进行高效的自由能计算。与之前的加速分子动力学(aMD)一样,GaMD允许进行“无约束”的增强采样,无需设置预定义的集体变量,因此对于研究复杂的生物分子构象变化(如蛋白质折叠和配体结合)很有用。此外,由于在GaMD中使用遵循高斯分布的谐波函数构建增强势,因此可以应用二阶累积量展开来恢复蛋白质和其他大型生物分子的原始自由能分布,这解决了先前aMD方法长期存在的能量重加权问题。综上所述,GaMD在大型生物分子的无约束增强采样和自由能计算方面都具有主要优势。在这里,我们在现有aMD功能的基础上,在NAMD软件包中实现了GaMD,并在三个模型系统上进行了验证:丙氨酸二肽、奇果菌素快速折叠蛋白和M型毒蕈碱G蛋白偶联受体(GPCR)。对于丙氨酸二肽,虽然进行30 ns的传统分子动力学(cMD)模拟收敛性很差,但相同长度的GaMD模拟产生的自由能分布与1000 ns cMD模拟的自由能分布在数量上一致。进一步的GaMD模拟捕捉到了奇果菌素的折叠以及内源性激动剂乙酰胆碱(ACh)与M型毒蕈碱受体的结合。重新加权的自由能分布用于定量表征蛋白质折叠和配体结合途径。在可扩展的NAMD中实现的GaMD广泛适用于大型生物分子的增强采样和自由能计算。