Department of Informatics, Bioinformatics and Computational Biology - i12, TUM-Technical University of Munich, Boltzmannstr. 3, Garching, 85748, Munich, Germany.
TUM Graduate School, Center of Doctoral Studies in Informatics and its Applications (CeDoSIA), Boltzmannstr. 11, 85748, Garching, Germany.
Hum Genet. 2022 Oct;141(10):1629-1647. doi: 10.1007/s00439-021-02411-y. Epub 2021 Dec 30.
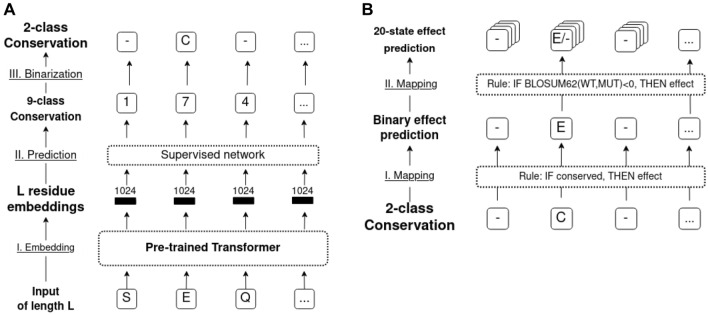
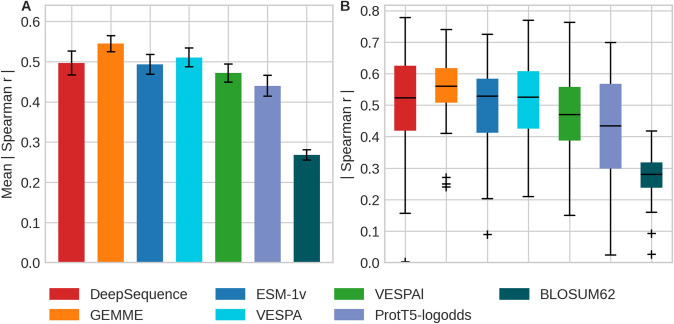
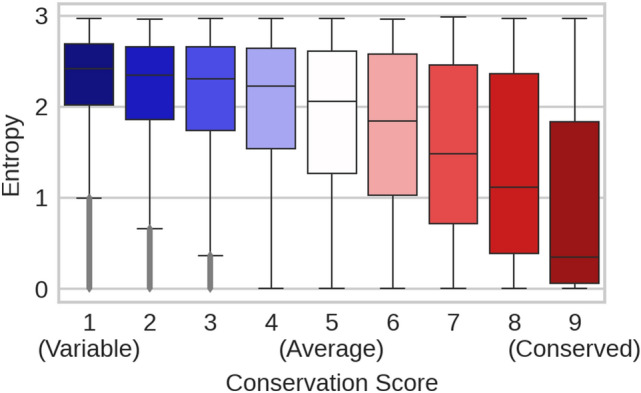
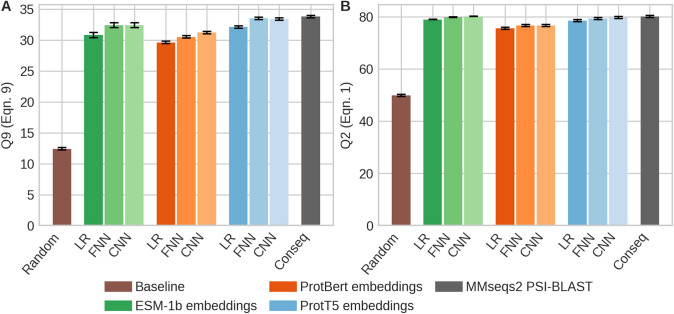
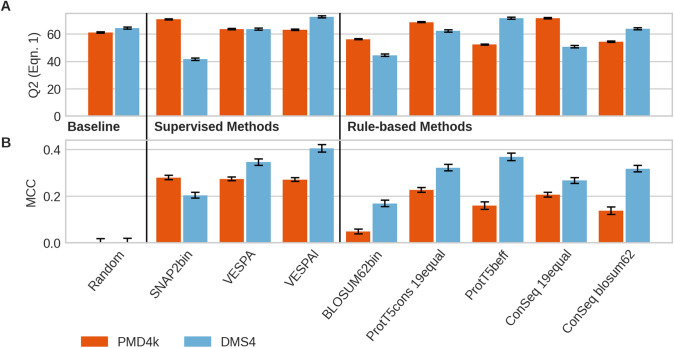
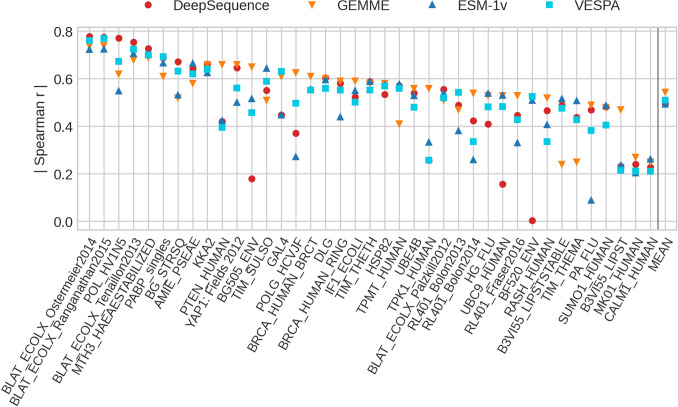
The emergence of SARS-CoV-2 variants stressed the demand for tools allowing to interpret the effect of single amino acid variants (SAVs) on protein function. While Deep Mutational Scanning (DMS) sets continue to expand our understanding of the mutational landscape of single proteins, the results continue to challenge analyses. Protein Language Models (pLMs) use the latest deep learning (DL) algorithms to leverage growing databases of protein sequences. These methods learn to predict missing or masked amino acids from the context of entire sequence regions. Here, we used pLM representations (embeddings) to predict sequence conservation and SAV effects without multiple sequence alignments (MSAs). Embeddings alone predicted residue conservation almost as accurately from single sequences as ConSeq using MSAs (two-state Matthews Correlation Coefficient-MCC-for ProtT5 embeddings of 0.596 ± 0.006 vs. 0.608 ± 0.006 for ConSeq). Inputting the conservation prediction along with BLOSUM62 substitution scores and pLM mask reconstruction probabilities into a simplistic logistic regression (LR) ensemble for Variant Effect Score Prediction without Alignments (VESPA) predicted SAV effect magnitude without any optimization on DMS data. Comparing predictions for a standard set of 39 DMS experiments to other methods (incl. ESM-1v, DeepSequence, and GEMME) revealed our approach as competitive with the state-of-the-art (SOTA) methods using MSA input. No method outperformed all others, neither consistently nor statistically significantly, independently of the performance measure applied (Spearman and Pearson correlation). Finally, we investigated binary effect predictions on DMS experiments for four human proteins. Overall, embedding-based methods have become competitive with methods relying on MSAs for SAV effect prediction at a fraction of the costs in computing/energy. Our method predicted SAV effects for the entire human proteome (~ 20 k proteins) within 40 min on one Nvidia Quadro RTX 8000. All methods and data sets are freely available for local and online execution through bioembeddings.com, https://github.com/Rostlab/VESPA , and PredictProtein.
SARS-CoV-2 变体的出现强调了需要有工具来解释单个氨基酸变异 (SAV) 对蛋白质功能的影响。虽然深度突变扫描 (DMS) 集继续扩展我们对单个蛋白质突变景观的理解,但结果继续挑战分析。蛋白质语言模型 (pLM) 使用最新的深度学习 (DL) 算法利用不断增长的蛋白质序列数据库。这些方法学会从整个序列区域的上下文预测缺失或屏蔽的氨基酸。在这里,我们使用 pLM 表示 (嵌入) 来预测序列保守性和 SAV 效应,而无需进行多重序列比对 (MSA)。仅嵌入就可以从单个序列中几乎与使用 MSA 的 ConSeq 一样准确地预测残基保守性 (对于 ProtT5 嵌入,两状态马修斯相关系数-MCC-为 0.596±0.006 与 ConSeq 的 0.608±0.006)。将保守性预测与 BLOSUM62 替换分数以及 pLM 掩蔽重建概率一起输入到无需比对的变体效应得分预测的简单逻辑回归 (LR) 集成中 (VESPA) 无需对 DMS 数据进行任何优化即可预测 SAV 效应幅度。将一组 39 个 DMS 实验的标准集与其他方法 (包括 ESM-1v、DeepSequence 和 GEMME) 的预测进行比较,结果表明,我们的方法与使用 MSA 输入的最先进 (SOTA) 方法具有竞争力。没有一种方法始终优于所有其他方法,无论是在应用的性能度量上,还是在统计上都没有显著差异。最后,我们研究了四种人类蛋白质的 DMS 实验的二元效应预测。总体而言,基于嵌入的方法已经在 SAV 效应预测方面与依赖 MSA 的方法竞争,其计算/能量成本只是一小部分。我们的方法在一台 Nvidia Quadro RTX 8000 上仅需 40 分钟即可预测整个人类蛋白质组 (~20k 个蛋白质) 的 SAV 效应。所有方法和数据集均可通过 bioembeddings.com、https://github.com/Rostlab/VESPA 和 PredictProtein 免费用于本地和在线执行。