Varukolu Mahipal, Palnati Manojkumar, Nampally Venkatesh, Gangadhari Suresh, Vadluri Manaiah, Tigulla Parthasarathy
Department of Chemistry, Osmania University, Hyderabad 500007 Telangana, India.
ACS Omega. 2021 Dec 22;7(1):810-822. doi: 10.1021/acsomega.1c05464. eCollection 2022 Jan 11.
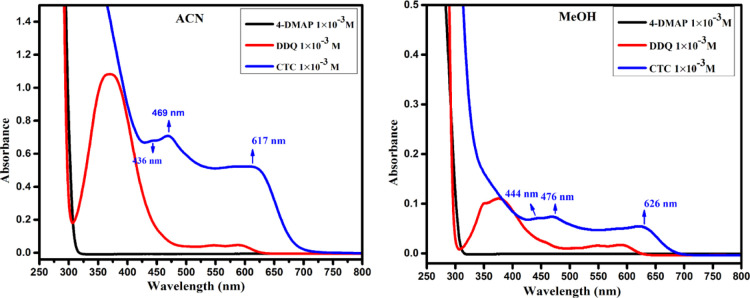
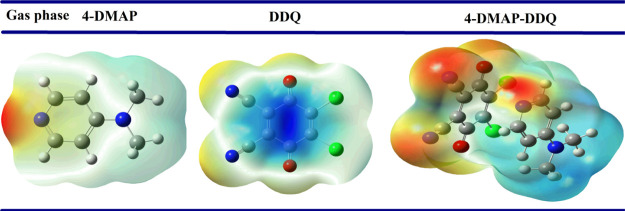
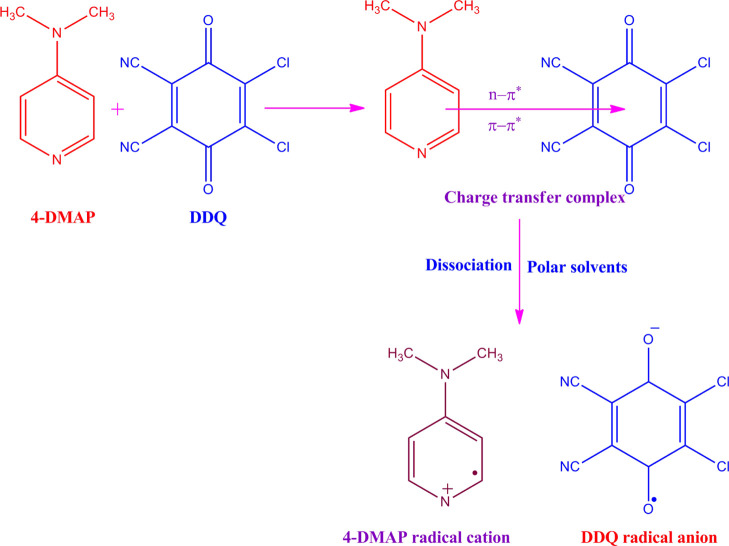
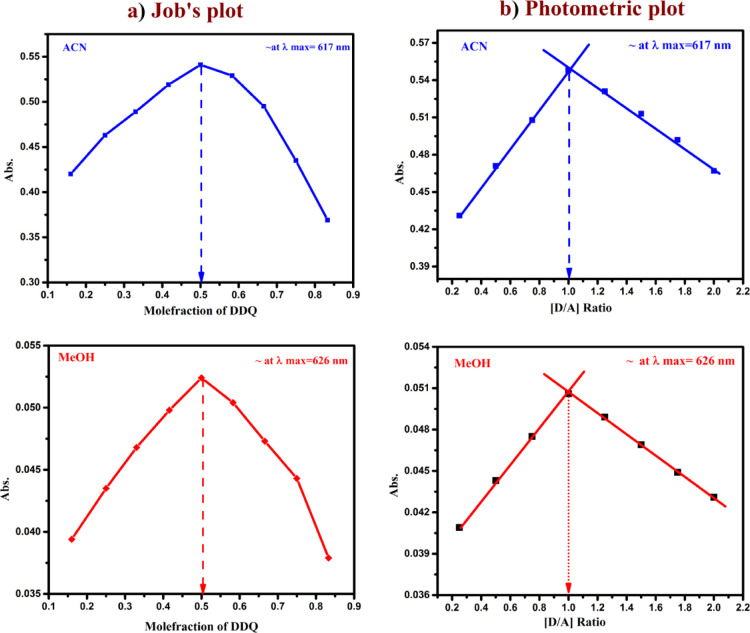
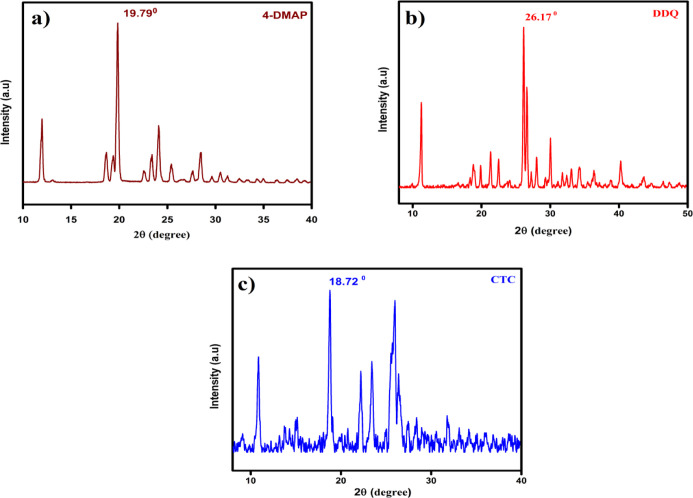
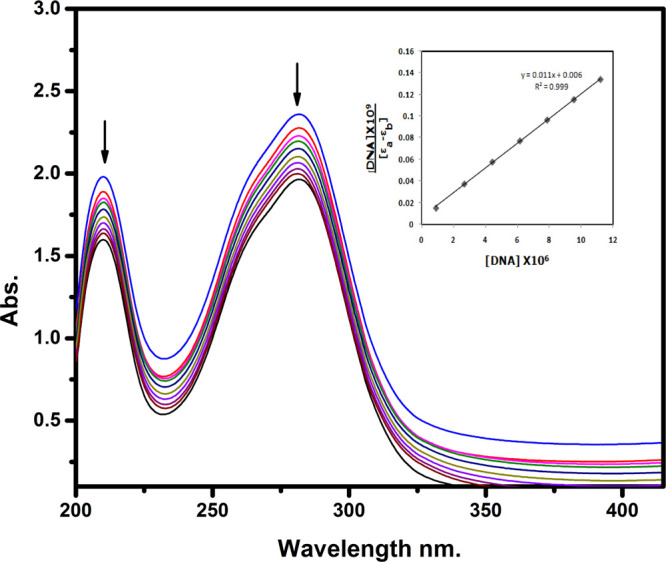
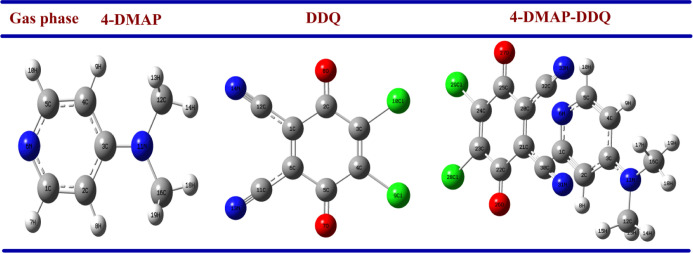
A combined experimental and theoretical study of the electron donor 4-dimethylaminopyridine (4-DMAP) with the electron acceptor 2, 3-dichloro-5, 6-dicyano--benzoquinone (DDQ) has been made in acetonitrile (ACN) and methanol (MeOH) media at room temperature. The stoichiometry proportion of the charge transfer (CT) complex was determined using Job's and photometric titration methods and found to be 1:1. The association constant ( ), molar absorptivity (ε), and spectroscopic physical parameters were used to know the stability of the CT complex. The CT complex shows maximum stability in a high-polar solvent (ACN) compared to a less-polar solvent (MeOH). The prepared complex was characterized by Fourier transform infrared, NMR, powder X-ray diffraction, and scanning electron microscopy-energy-dispersive X-ray analysis. The nature of DNA binding ability of the complex was probed using UV-visible spectroscopy, and the binding mode of the CT complex is intercalative. The intrinsic binding constant ( ) value is 1.8 × 10 M. It reveals a primary indication for developing a pharmaceutical drug in the future due to its high binding affinity with the CT complex. The theoretical study was carried out by density functional theory (DFT), and the basis set is wB97XD/6-31G(d,p), with gas-phase and PCM analysis, which supports experimental results. Natural atomic charges, state dipole moments, electron density difference maps, reactivity parameters, and FMO surfaces were also evaluated. The MEP maps indicate the electrophilic nature of DDQ and the nucleophilic nature of 4-DMAP. The electronic spectrum computed using time-dependent DFT (TD-DFT) via a polarizable continuum salvation approach, PCM/TD-DFT, along with natural transition orbital analysis is fully correlated with the experimental outcomes.
在室温下,于乙腈(ACN)和甲醇(MeOH)介质中,对电子供体4 - 二甲氨基吡啶(4 - DMAP)与电子受体2,3 - 二氯 - 5,6 - 二氰基 - 对苯醌(DDQ)进行了实验与理论相结合的研究。采用乔布氏法和光度滴定法确定了电荷转移(CT)配合物的化学计量比,发现其为1:1。利用缔合常数( )、摩尔吸光系数(ε)以及光谱物理参数来了解CT配合物的稳定性。与极性较小的溶剂(MeOH)相比,CT配合物在高极性溶剂(ACN)中表现出最大稳定性。通过傅里叶变换红外光谱、核磁共振、粉末X射线衍射以及扫描电子显微镜 - 能量色散X射线分析对所制备的配合物进行了表征。利用紫外 - 可见光谱探究了该配合物与DNA结合能力的性质,CT配合物的结合模式为插入式。固有结合常数( )值为1.8×10 M。由于其与CT配合物具有高结合亲和力,这为未来开发药物提供了初步迹象。理论研究采用密度泛函理论(DFT)进行,基组为wB97XD/6 - 31G(d,p),并进行了气相和PCM分析,这支持了实验结果。还评估了自然原子电荷、态偶极矩、电子密度差图、反应性参数以及前线分子轨道表面。分子静电势(MEP)图表明了DDQ的亲电性质和4 - DMAP的亲核性质。通过极化连续介质溶剂化方法PCM/TD - DFT,利用含时密度泛函理论(TD - DFT)计算的电子光谱以及自然跃迁轨道分析与实验结果完全相关。