Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, Shanghai, China.
Cancer Med. 2022 Jun;11(11):2244-2258. doi: 10.1002/cam4.4547. Epub 2022 Jan 31.
Single-cell transcriptomics has been used to investigate various tumors to elucidate the molecular distinction of all cell type compositions of a complex mix.
This study aimed to investigate malignant-cell-specific genes to explore diagnostic and therapeutic biomarkers using single-cell transcriptomic data of lung adenocarcinoma.
MATERIALS & METHODS: 10X single-cell RNA-seq data of fourteen patients with lung adenocarcinoma were analyzed. Genes that expressed differentially and those with higher confidence to distinguish tumor cells from normal cells were picked out using the ROC curves. The LASSO regression method was used to select most markedly correlated genes to predict the malignancy of every single cell within a model. We also conducted further experiments to determine their roles in lung cancer in vitro.
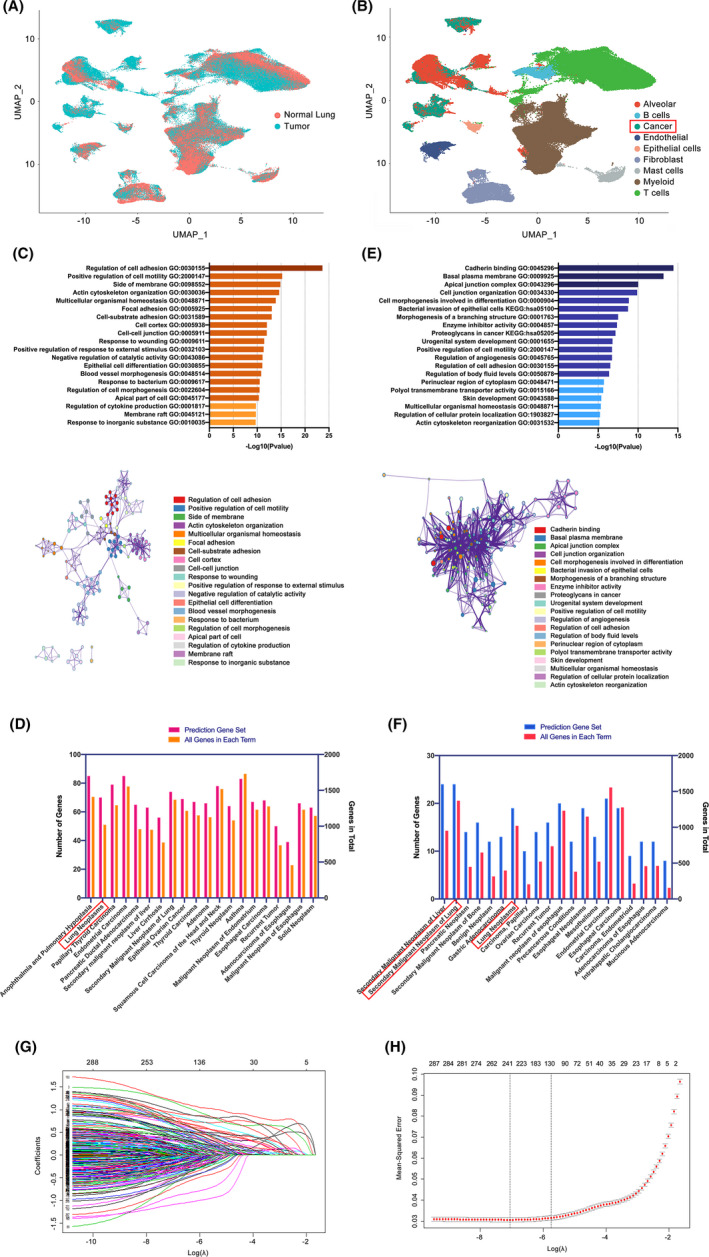
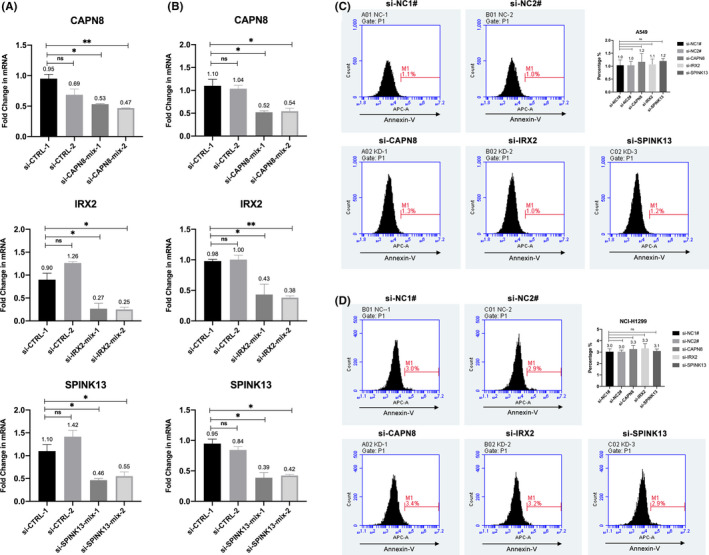
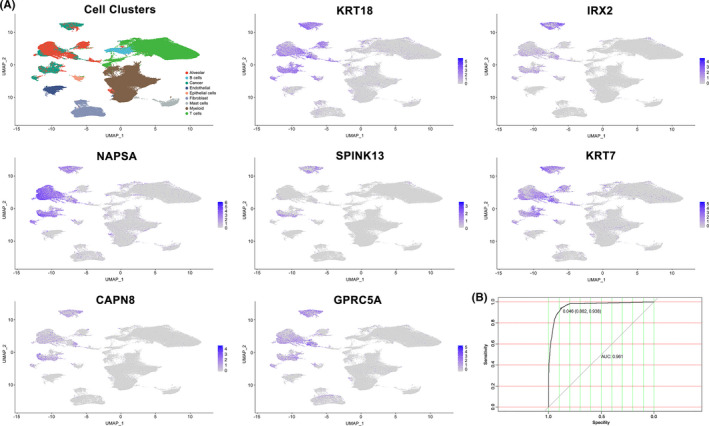
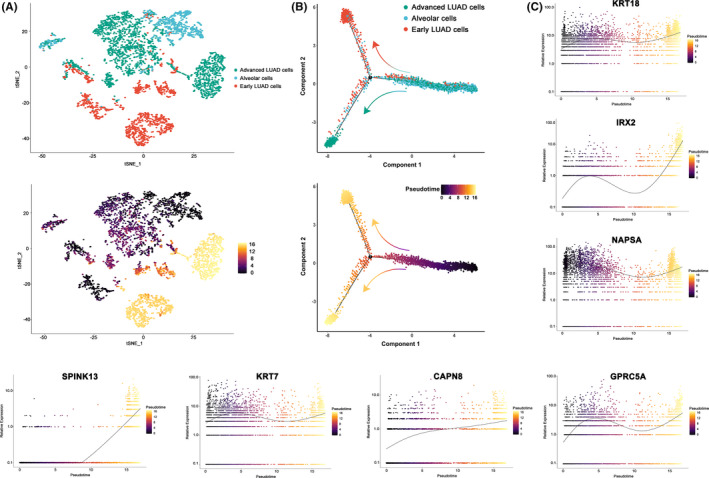
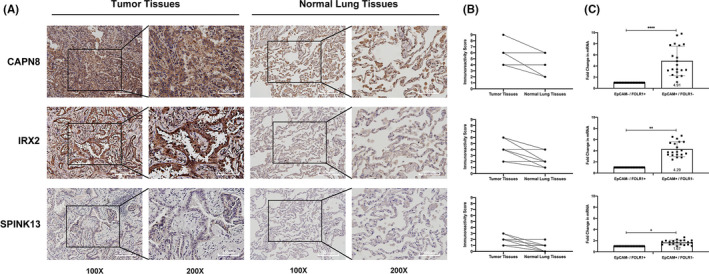
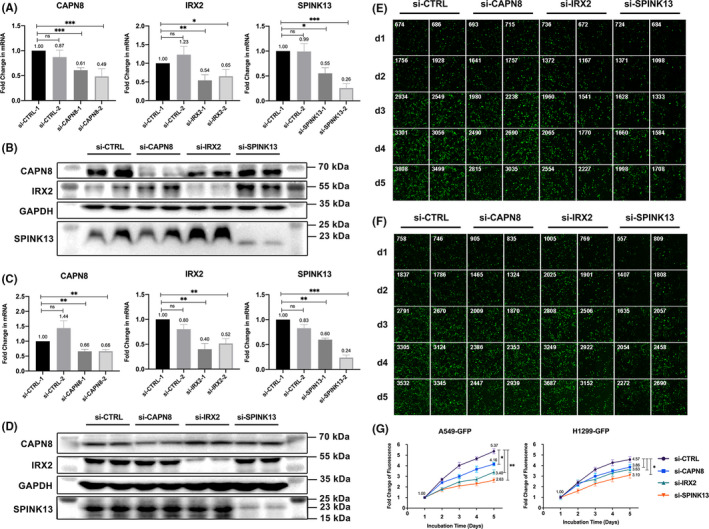
Twenty two thousand four hundred and ninety one tumor and 181 666 normal single cells were analyzed where 369 genes were found to be specifically expressed in single malignant cells. Seventy of them, encoding secreted or membrane-bound proteins, showed involvement in cell-to-cell communications in tumor biology. KRT18 and the other six genes were identified as predictors to distinguish single malignant cells and were integrated to construct an accurate (96.1%) predicting model. Notably, IRX2, SPINK13, and CAPN8 outperformed the other four genes. Further experiments confirmed the upregulation of them in lung adenocarcinoma at both tissue and cell levels. Proliferative capacities of lung adenocarcinoma cells were attenuated by knocking-down of either of them. However, targeting CAPN8, IRX2, or SPINK13 hardly exerted a cytotoxic effect on these cells.
Apart from the current model, similar tools were still warranted using single-cell RNA-seq data of more types of tumors. The three genes identified as potential therapeutic targets in the present study still need to be validated in more in lung cancer.
Our model can aid the analyses of single-cell sequencing data. CAPN8, IRX2, and SPINK13 may serve as novel targets of targeted and immune-based therapies in lung adenocarcinoma.
单细胞转录组学已被用于研究各种肿瘤,以阐明复杂混合物中所有细胞类型组成的分子差异。
本研究旨在使用肺腺癌的单细胞转录组学数据,研究恶性细胞特异性基因,以探索诊断和治疗生物标志物。
对 14 例肺腺癌患者的 10X 单细胞 RNA-seq 数据进行分析。使用 ROC 曲线筛选出差异表达且置信度更高的基因,以区分肿瘤细胞和正常细胞。LASSO 回归方法用于选择与模型中每个单细胞的恶性程度最相关的基因进行预测。我们还进行了进一步的实验,以确定它们在体外肺癌中的作用。
分析了 24910 个肿瘤和 181666 个正常单细胞,发现 369 个基因在单个恶性细胞中特异性表达。其中 70 个基因编码分泌或膜结合蛋白,它们在肿瘤生物学中的细胞间通讯中发挥作用。KRT18 和其他六个基因被确定为区分单个恶性细胞的预测因子,并整合到一个准确的(96.1%)预测模型中。值得注意的是,IRX2、SPINK13 和 CAPN8 的表现优于其他四个基因。进一步的实验证实,它们在肺腺癌组织和细胞水平上均上调。敲低其中任何一个基因都能减弱肺腺癌细胞的增殖能力。然而,针对 CAPN8、IRX2 或 SPINK13 几乎对这些细胞没有细胞毒性作用。
除了当前的模型外,使用更多类型肿瘤的单细胞 RNA-seq 数据仍需要类似的工具。本研究中鉴定的三个基因作为潜在的治疗靶点仍需要在更多的肺癌中进行验证。
我们的模型可以辅助单细胞测序数据的分析。CAPN8、IRX2 和 SPINK13 可能成为肺腺癌靶向和免疫治疗的新靶点。