Nipun Tanzina Sharmin, Ema Tanzila Ismail, Mia Md Abdur Rashid, Hossen Md Saddam, Arshe Farzana Alam, Ahmed Shahlaa Zernaz, Masud Afsana, Taheya Fatiha Faheem, Khan Arysha Alif, Haque Fauzia, Azad Salauddin Al, Al Hasibuzzaman Md, Tanbir Mohammad, Anis Samin, Akter Sharmin, Mily Sabrina Jahan, Dey Dipta
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, International Islamic University Malaysia, Kuantan, Malaysia.
Department of Biochemistry and Microbiology, North South University, Dhaka, Bangladesh.
J Adv Vet Anim Res. 2021 Sep 29;8(4):540-556. doi: 10.5455/javar.2021.h544. eCollection 2021 Dec.
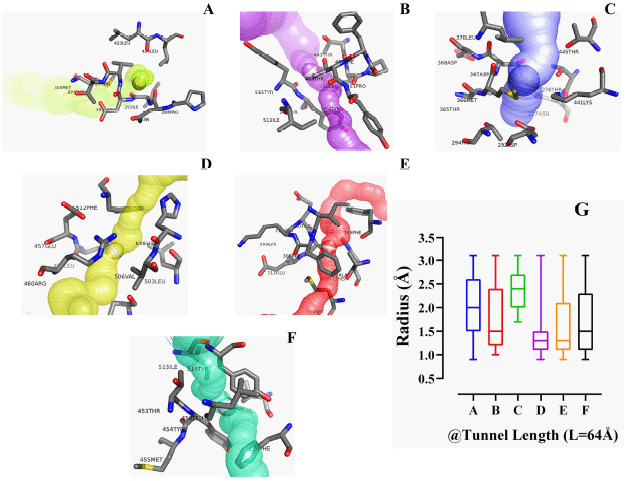
This research aims to study the target specificity of selective bioactive compounds in complexing with the human angiotensin-converting enzyme (hACE2) receptor to impede the severe acute respiratory syndrome coronavirus 2 influx mechanism resulting in cardiac injury and depending on the receptor's active site properties and quantum tunneling.
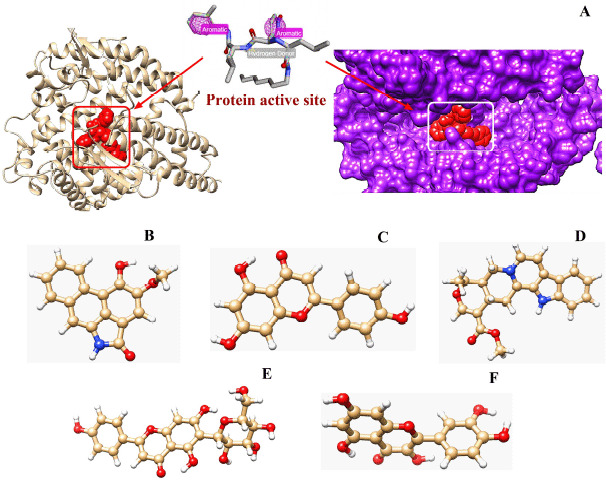
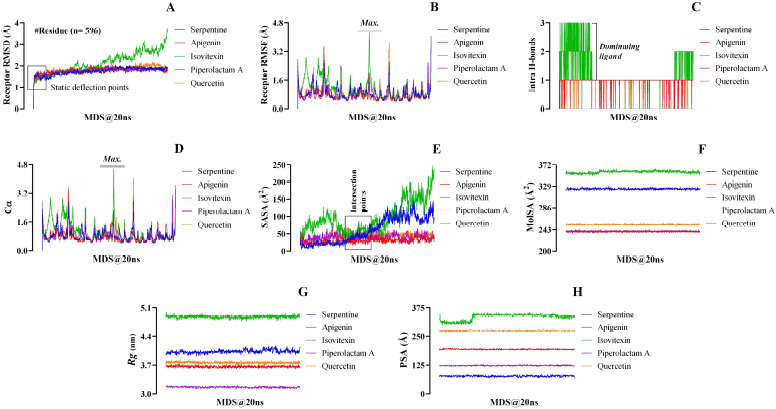
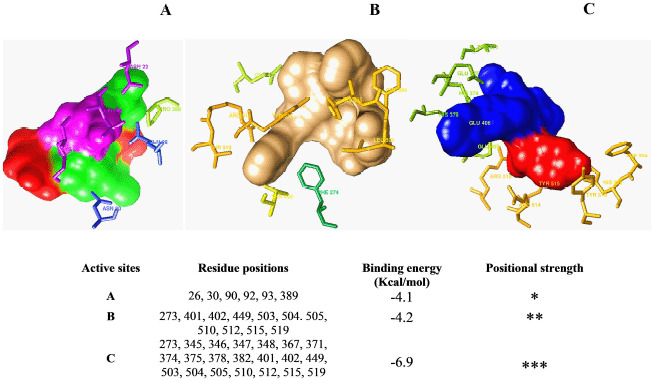
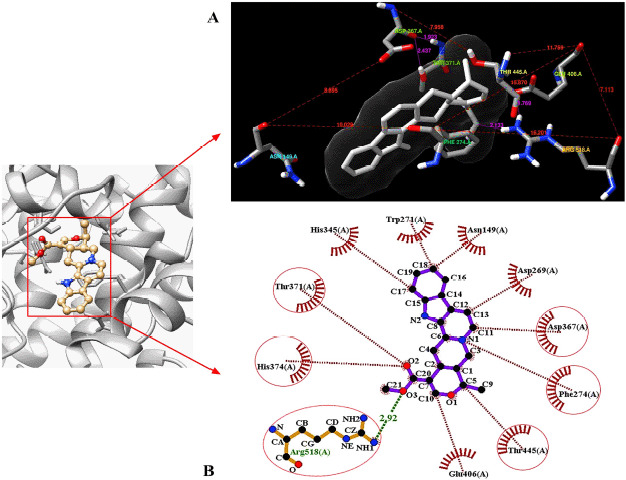
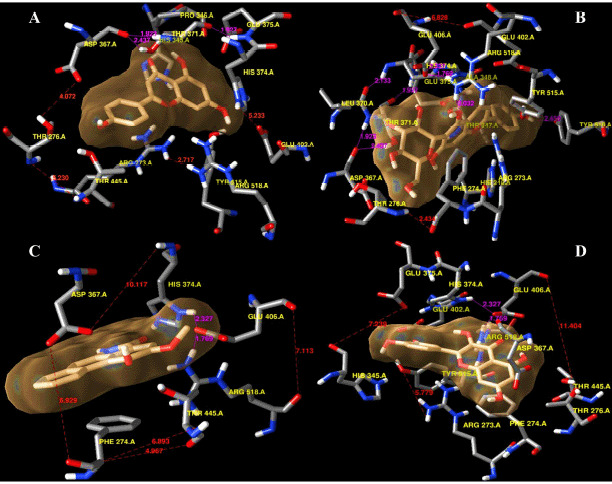
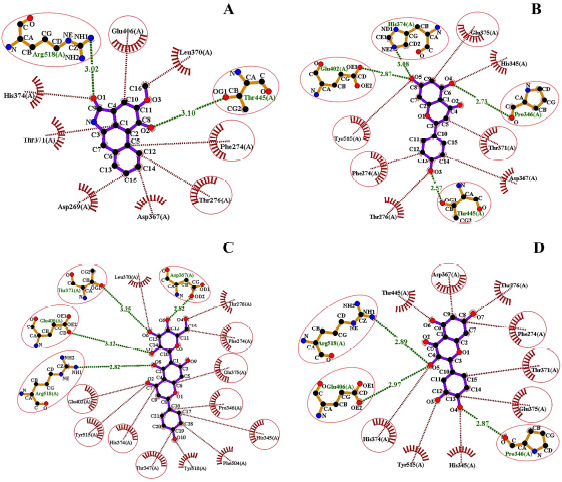
A library of 120 phytochemical ligands was prepared, from which 5 were selected considering their absorption, distribution, metabolism, and excretion (ADMET) and quantitative structure-activity relationship (QSAR) profiles. The protein active sites and belonging quantum tunnels were defined to conduct supramolecular docking of the aforementioned ligands. The hydrogen bond formation and hydrophobic interactions between the ligand-receptor complexes were studied following the molecular docking steps. A comprehensive molecular dynamic simulation (MDS) was conducted for each of the ligand-receptor complexes to figure out the values - root mean square deviation (RMSD) (Å), root mean square fluctuation (RMSF) (Å), H-bonds, Cα, solvent accessible surface area (SASA) (Å), molecular surface area (MolSA) (Å), Rg (nm), and polar surface area (PSA) (Å). Finally, computational programming and algorithms were used to interpret the dynamic simulation outputs into their graphical quantitative forms.
ADMET and QSAR profiles revealed that the most active candidates from the library to be used were apigenin, isovitexin, piperolactam A, and quercetin as test ligands, whereas serpentine as the control. Based on the binding affinities of supramolecular docking and the parameters of molecular dynamic simulation, the strength of the test ligands can be classified as isovitexin > quercetin > piperolactam A > apigenin when complexed with the hACE2 receptor. Surprisingly, serpentine showed lower affinity (-8.6 kcal/mol) than that of isovitexin (-9.9 kcal/mol) and quercetin (-8.9 kcal/mol). The MDS analysis revealed all ligands except isovitexin having a value lower than 2.5 Ǻ. All the test ligands exhibited acceptable fluctuation ranges of RMSD (Å), RMSF (Å), H-bonds, Cα, SASA (Å), MolSA (Å), Rg (nm), and PSA (Å) values.
Considering each of the parameters of molecular optimization, docking, and dynamic simulation interventions, all of the test ligands can be suggested as potential targeted drugs in blocking the hACE2 receptor.
本研究旨在探讨选择性生物活性化合物与人类血管紧张素转换酶(hACE2)受体结合的靶点特异性,以阻止严重急性呼吸综合征冠状病毒2的内流机制,该机制导致心脏损伤,并取决于受体的活性位点特性和量子隧穿。
制备了一个包含120种植物化学配体的文库,根据其吸收、分布、代谢和排泄(ADMET)以及定量构效关系(QSAR)图谱从中选择了5种。定义了蛋白质活性位点和所属的量子隧道,以对上述配体进行超分子对接。按照分子对接步骤研究了配体-受体复合物之间的氢键形成和疏水相互作用。对每个配体-受体复合物进行了全面的分子动力学模拟(MDS),以确定均方根偏差(RMSD)(埃)、均方根波动(RMSF)(埃)、氢键、Cα、溶剂可及表面积(SASA)(埃)、分子表面积(MolSA)(埃)、Rg(纳米)和极性表面积(PSA)(埃)的值。最后,使用计算编程和算法将动态模拟输出解释为图形定量形式。
ADMET和QSAR图谱显示,文库中最具活性的候选物为芹菜素、异荭草素、胡椒内酰胺A和槲皮素作为测试配体,而蛇根碱作为对照。基于超分子对接的结合亲和力和分子动力学模拟参数,当与hACE2受体结合时,测试配体的强度可分类为异荭草素>槲皮素>胡椒内酰胺A>芹菜素。令人惊讶的是,蛇根碱的亲和力(-8.6千卡/摩尔)低于异荭草素(-9.9千卡/摩尔)和槲皮素(-8.9千卡/摩尔)。MDS分析显示,除异荭草素外,所有配体的值均低于2.5埃。所有测试配体的RMSD(埃)、RMSF(埃)、氢键、Cα、SASA(埃)、MolSA(埃)、Rg(纳米)和PSA(埃)值均表现出可接受的波动范围。
考虑到分子优化、对接和动态模拟干预的各项参数,所有测试配体均可被建议作为阻断hACE2受体的潜在靶向药物。