Department of Rheumatology, Leiden University Medical Center, Leiden, The Netherlands.
Leiden Computational Biology Center, Leiden University Medical Center, Leiden, The Netherlands.
J Am Med Inform Assoc. 2022 Apr 13;29(5):761-769. doi: 10.1093/jamia/ocac008.
To facilitate patient disease subset and risk factor identification by constructing a pipeline which is generalizable, provides easily interpretable results, and allows replication by overcoming electronic health records (EHRs) batch effects.
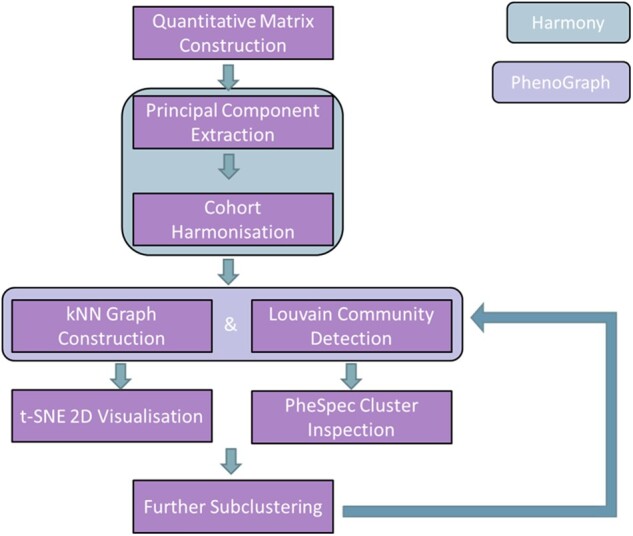
We used 1872 billing codes in EHRs of 102 880 patients from 12 healthcare systems. Using tools borrowed from single-cell omics, we mitigated center-specific batch effects and performed clustering to identify patients with highly similar medical history patterns across the various centers. Our visualization method (PheSpec) depicts the phenotypic profile of clusters, applies a novel filtering of noninformative codes (Ranked Scope Pervasion), and indicates the most distinguishing features.
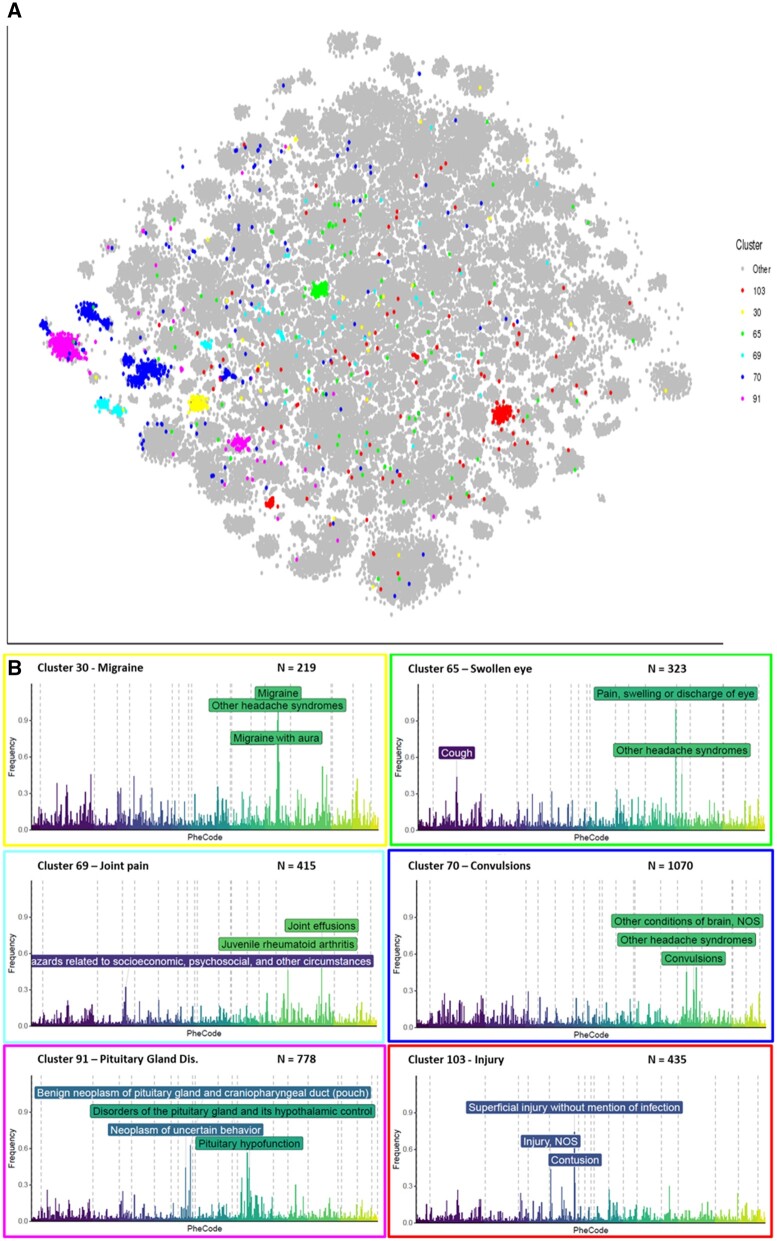
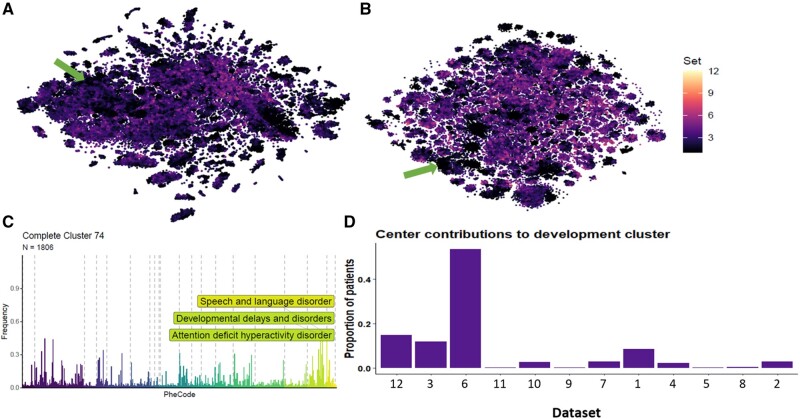
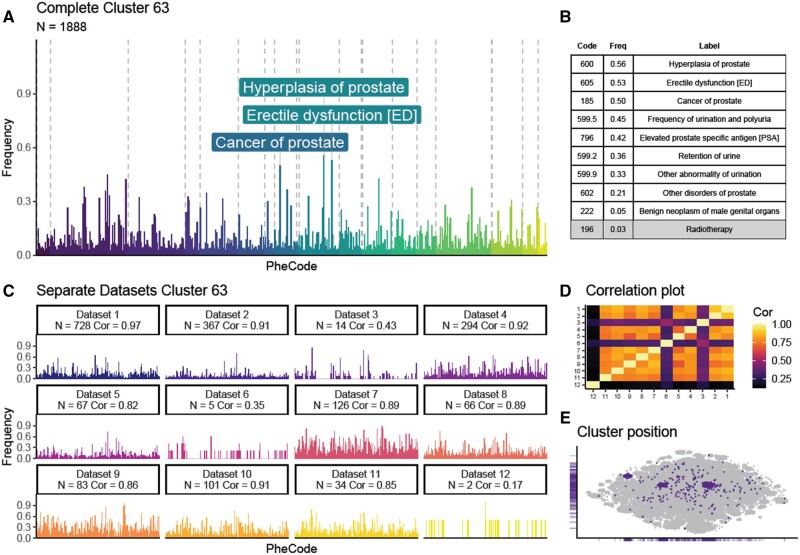
We observed 114 clinically meaningful profiles, for example, linking prostate hyperplasia with cancer and diabetes with cardiovascular problems and grouping pediatric developmental disorders. Our framework identified disease subsets, exemplified by 6 "other headache" clusters, where phenotypic profiles suggested different underlying mechanisms: migraine, convulsion, injury, eye problems, joint pain, and pituitary gland disorders. Phenotypic patterns replicated well, with high correlations of ≥0.75 to an average of 6 (2-8) of the 12 different cohorts, demonstrating the consistency with which our method discovers disease history profiles.
Costly clinical research ventures should be based on solid hypotheses. We repurpose methods from single-cell omics to build these hypotheses from observational EHR data, distilling useful information from complex data.
We establish a generalizable pipeline for the identification and replication of clinically meaningful (sub)phenotypes from widely available high-dimensional billing codes. This approach overcomes datatype problems and produces comprehensive visualizations of validation-ready phenotypes.
通过构建一个可推广、提供易于解释的结果且可克服电子健康记录(EHR)批次效应的管道,方便患者疾病亚组和危险因素的识别。
我们使用了来自 12 个医疗系统的 102880 名患者的 1872 个计费代码。使用单细胞组学借来的工具,我们减轻了中心特定的批次效应,并进行聚类以识别在各个中心具有高度相似病史模式的患者。我们的可视化方法(PheSpec)描绘了聚类的表型特征,应用了一种新的非信息性代码过滤方法(排名范围普遍),并指出了最具区别性的特征。
我们观察到了 114 个具有临床意义的特征,例如将前列腺增生与癌症以及糖尿病与心血管问题联系起来,并将儿科发育障碍分组。我们的框架确定了疾病亚组,例如 6 个“其他头痛”聚类,其表型特征表明了不同的潜在机制:偏头痛、抽搐、损伤、眼部问题、关节痛和垂体腺疾病。表型模式复制得很好,相关性高达 0.75 以上,平均与 12 个不同队列中的 6 个(2-8 个)相关,这表明了我们的方法发现疾病史特征的一致性。
昂贵的临床研究应该基于可靠的假设。我们从单细胞组学中重新利用方法,从观察性 EHR 数据中构建这些假设,从复杂的数据中提取有用的信息。
我们建立了一个可推广的管道,用于从广泛可用的高维计费代码中识别和复制具有临床意义的(亚)表型。这种方法克服了数据类型问题,并产生了可验证的表型的全面可视化。