Department of Epidemiology and Biostatistics, School of Public Health, Medical College of Soochow University, Suzhou, China.
Jiangsu Key Laboratory of Preventive and Translational Medicine for Geriatric Diseases, Medical College of Soochow University, Suzhou, China.
Front Immunol. 2022 Jan 24;12:746998. doi: 10.3389/fimmu.2021.746998. eCollection 2021.
Growing evidence has shown that alterations in gut microbiota composition are associated with multiple autoimmune diseases (ADs). However, it is unclear whether these associations reflect a causal relationship.
To reveal the causal association between gut microbiota and AD, we conducted a two-sample Mendelian randomization (MR) analysis.
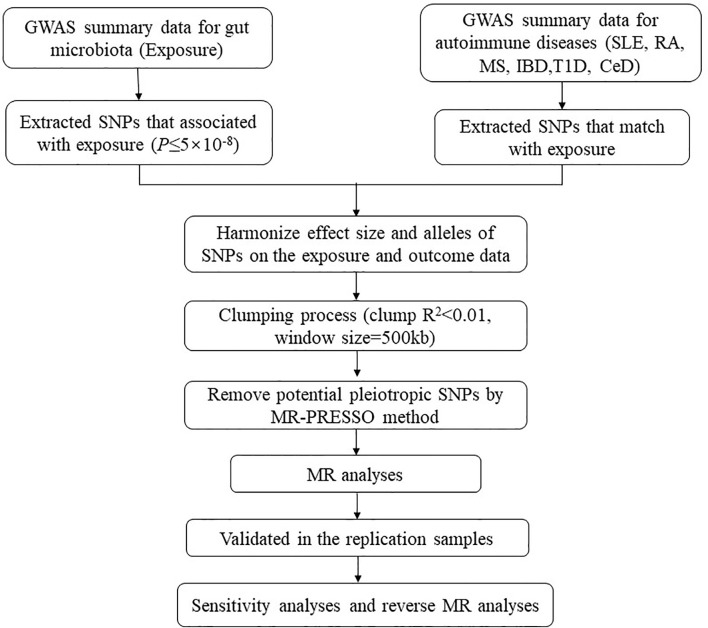
We assessed genome-wide association study (GWAS) summary statistics for gut microbiota and six common ADs, namely, systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, type 1 diabetes (T1D), and celiac disease (CeD), from published GWASs. Two-sample MR analyses were first performed to identify causal bacterial taxa for ADs in discovery samples. Significant bacterial taxa were further replicated in independent replication outcome samples. A series of sensitivity analyses was performed to validate the robustness of the results. Finally, a reverse MR analysis was performed to evaluate the possibility of reverse causation.
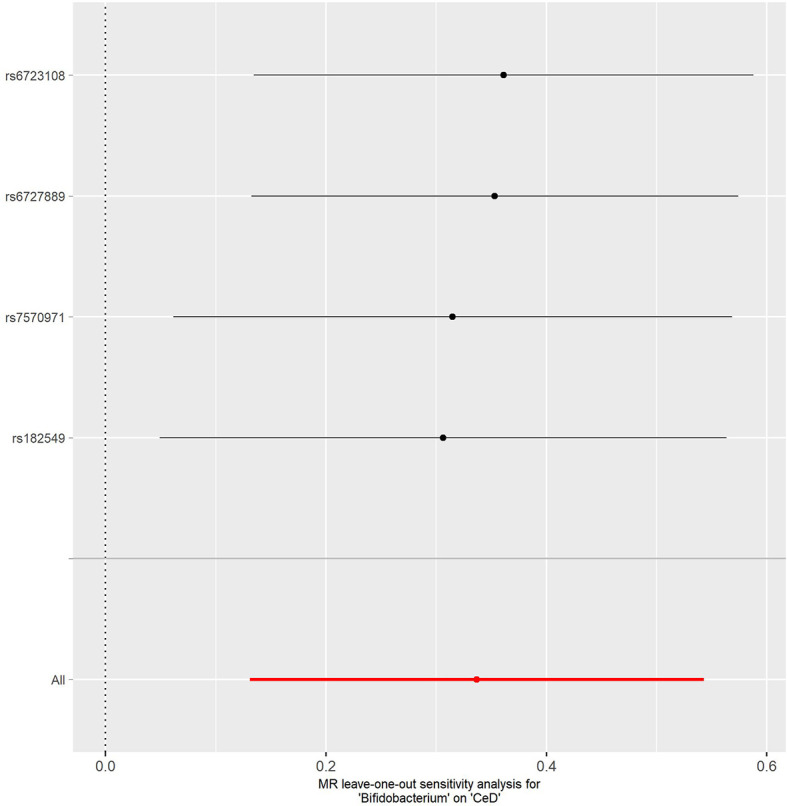
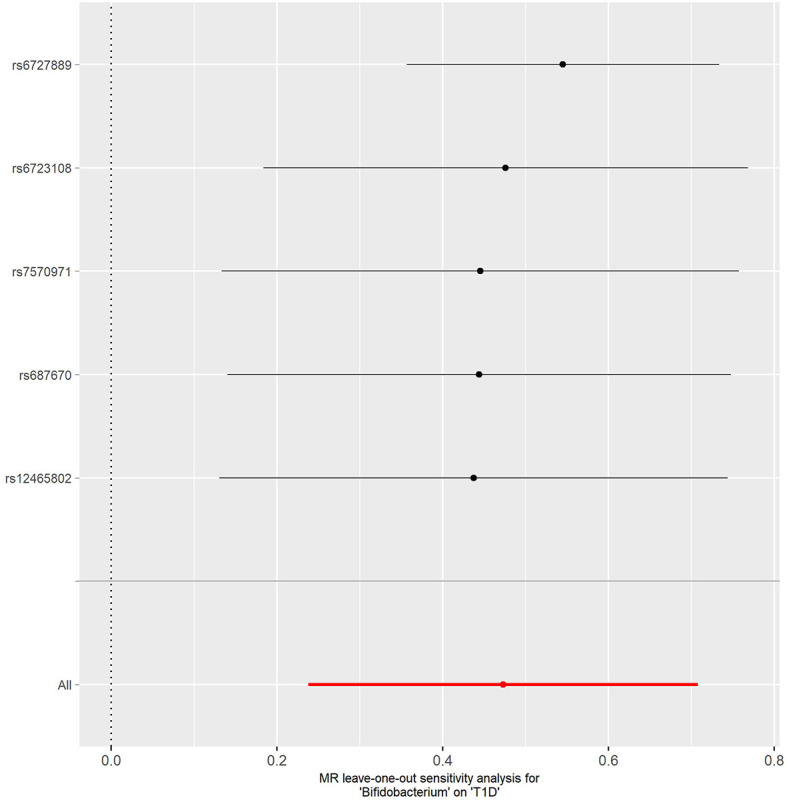
Combining the results from the discovery and replication stages, we identified one causal bacterial genus, . A higher relative abundance of the genus was associated with a higher risk of T1D [odds ratio (OR): 1.605; 95% CI, 1.339-1.922; = 4.19 × 10] and CeD (OR: 1.401; 95% CI, 1.139-1.722; = 2.03 × 10), respectively. Further sensitivity analyses validated the robustness of the above associations. The results of reverse MR analysis showed no evidence of reverse causality from T1D and CeD to the genus.
This study implied a causal relationship between the genus and T1D and CeD, thus providing novel insights into the gut microbiota-mediated development mechanism of ADs.
越来越多的证据表明,肠道微生物群落组成的改变与多种自身免疫性疾病(AD)有关。然而,目前尚不清楚这些关联是否反映了因果关系。
为了揭示肠道微生物群与 AD 之间的因果关系,我们进行了两样本孟德尔随机化(MR)分析。
我们评估了来自已发表 GWAS 的肠道微生物群和六种常见 AD(即系统性红斑狼疮、类风湿性关节炎、炎症性肠病、多发性硬化症、1 型糖尿病(T1D)和乳糜泻(CeD))的全基因组关联研究(GWAS)汇总统计数据。首先在发现样本中进行两样本 MR 分析,以确定 AD 的因果细菌分类群。在独立的复制结果样本中进一步复制显著的细菌分类群。进行了一系列敏感性分析以验证结果的稳健性。最后,进行了反向 MR 分析以评估反向因果关系的可能性。
结合发现和复制阶段的结果,我们确定了一个因果细菌属,.该属的相对丰度较高与 T1D 的风险增加相关[比值比(OR):1.605;95%可信区间,1.339-1.922; = 4.19 × 10]和 CeD(OR:1.401;95% CI,1.139-1.722; = 2.03 × 10),分别。进一步的敏感性分析验证了上述关联的稳健性。反向 MR 分析的结果表明,从 T1D 和 CeD 到 属没有反向因果关系的证据。
本研究暗示了 属与 T1D 和 CeD 之间存在因果关系,从而为 AD 的肠道微生物群介导的发病机制提供了新的见解。