Stringer Bas, de Ferrante Hans, Abeln Sanne, Heringa Jaap, Feenstra K Anton, Haydarlou Reza
Department of Computer Science, IBIVU-Center for Integrative Bioinformatics, Vrije Universiteit, 1081HV Amsterdam, The Netherlands.
Bioinformatics. 2022 Apr 12;38(8):2111-2118. doi: 10.1093/bioinformatics/btac071.
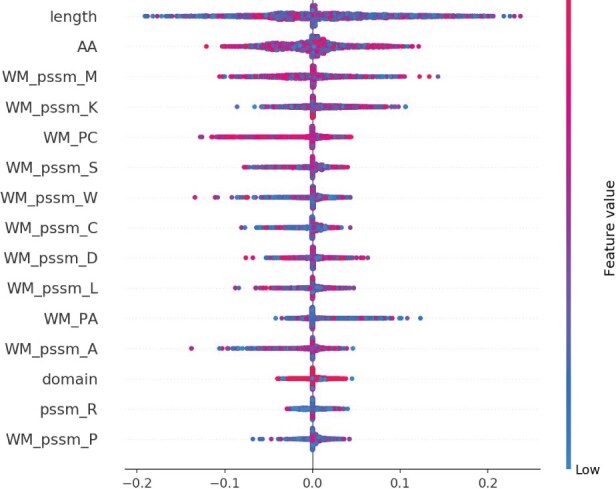
The interactions between proteins and other molecules are essential to many biological and cellular processes. Experimental identification of interface residues is a time-consuming, costly and challenging task, while protein sequence data are ubiquitous. Consequently, many computational and machine learning approaches have been developed over the years to predict such interface residues from sequence. However, the effectiveness of different Deep Learning (DL) architectures and learning strategies for protein-protein, protein-nucleotide and protein-small molecule interface prediction has not yet been investigated in great detail. Therefore, we here explore the prediction of protein interface residues using six DL architectures and various learning strategies with sequence-derived input features.
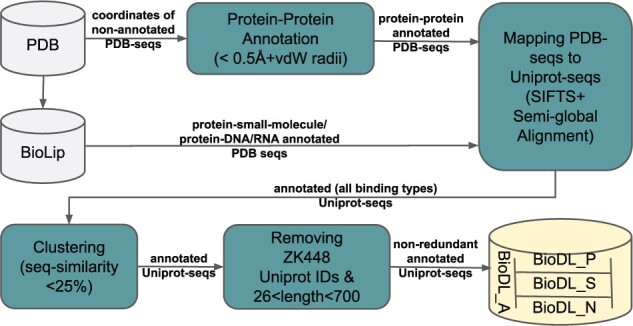
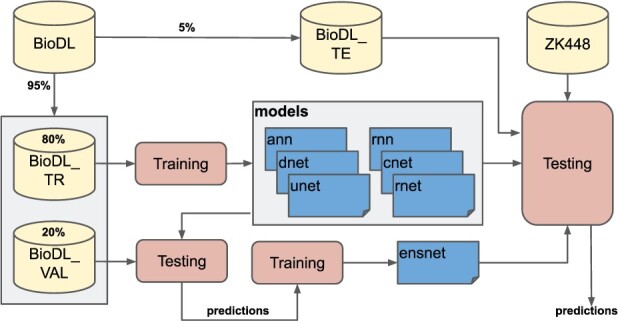
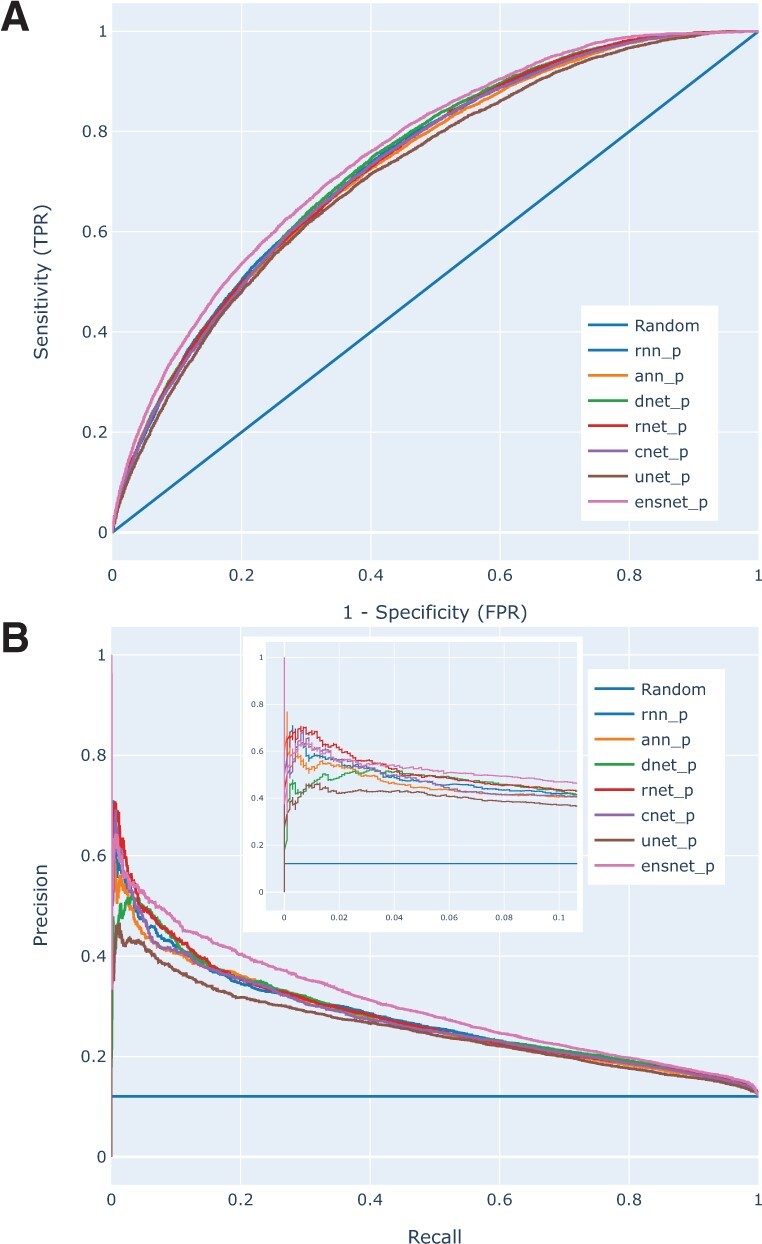
We constructed a large dataset dubbed BioDL, comprising protein-protein interactions from the PDB, and DNA/RNA and small molecule interactions from the BioLip database. We also constructed six DL architectures, and evaluated them on the BioDL benchmarks. This shows that no single architecture performs best on all instances. An ensemble architecture, which combines all six architectures, does consistently achieve peak prediction accuracy. We confirmed these results on the published benchmark set by Zhang and Kurgan (ZK448), and on our own existing curated homo- and heteromeric protein interaction dataset. Our PIPENN sequence-based ensemble predictor outperforms current state-of-the-art sequence-based protein interface predictors on ZK448 on all interaction types, achieving an AUC-ROC of 0.718 for protein-protein, 0.823 for protein-nucleotide and 0.842 for protein-small molecule.
Source code and datasets are available at https://github.com/ibivu/pipenn/.
Supplementary data are available at Bioinformatics online.
蛋白质与其他分子之间的相互作用对许多生物和细胞过程至关重要。通过实验鉴定界面残基是一项耗时、成本高且具有挑战性的任务,而蛋白质序列数据却无处不在。因此,多年来已经开发了许多计算和机器学习方法来从序列预测此类界面残基。然而,不同的深度学习(DL)架构和学习策略在蛋白质-蛋白质、蛋白质-核苷酸和蛋白质-小分子界面预测方面的有效性尚未得到详细研究。因此,我们在此使用六种DL架构和各种学习策略以及源自序列的输入特征来探索蛋白质界面残基的预测。
我们构建了一个名为BioDL的大型数据集,其中包括来自PDB的蛋白质-蛋白质相互作用以及来自BioLip数据库的DNA/RNA和小分子相互作用。我们还构建了六种DL架构,并在BioDL基准上对它们进行了评估。这表明没有一种架构在所有情况下都表现最佳。一种结合了所有六种架构的集成架构确实始终能达到峰值预测准确率。我们在Zhang和Kurgan(ZK448)发布的基准集以及我们自己现有的经过整理的同聚和异聚蛋白质相互作用数据集上证实了这些结果。我们基于序列的PIPENN集成预测器在ZK448上的所有相互作用类型上均优于当前基于序列的蛋白质界面预测器的最新技术水平,蛋白质-蛋白质相互作用的AUC-ROC为0.718,蛋白质-核苷酸相互作用的AUC-ROC为0.823,蛋白质-小分子相互作用的AUC-ROC为0.842。
源代码和数据集可在https://github.com/ibivu/pipenn/获取。
补充数据可在《生物信息学》在线获取。