CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, 200031, China.
University of Chinese Academy of Sciences, Beijing, 100049, China.
Genome Biol. 2022 Feb 28;23(1):62. doi: 10.1186/s13059-022-02627-9.
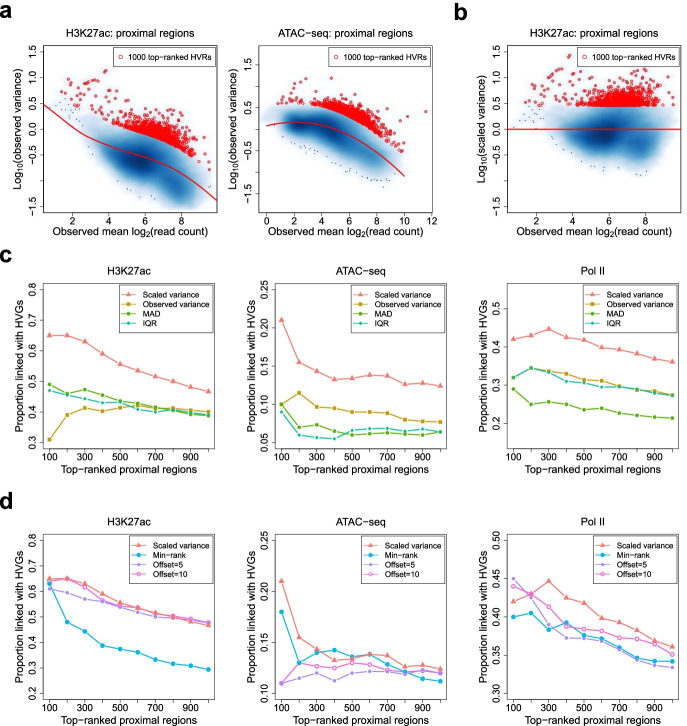
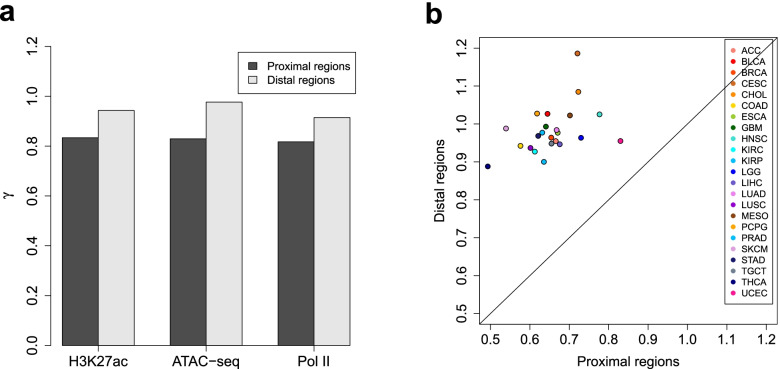
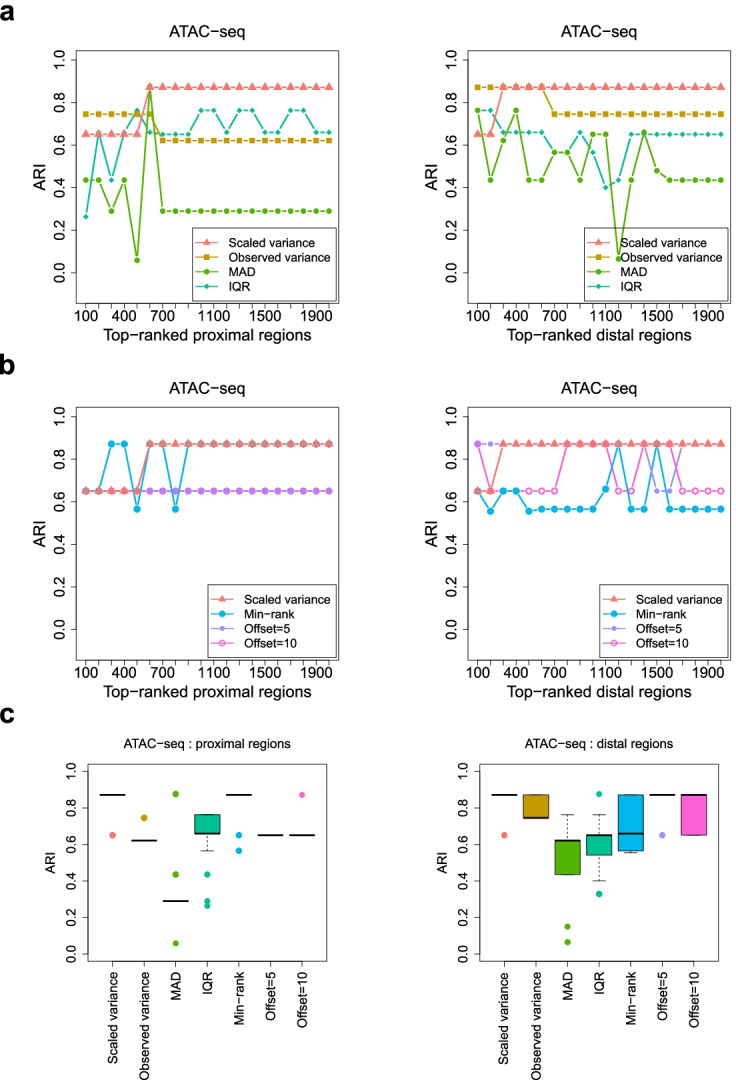
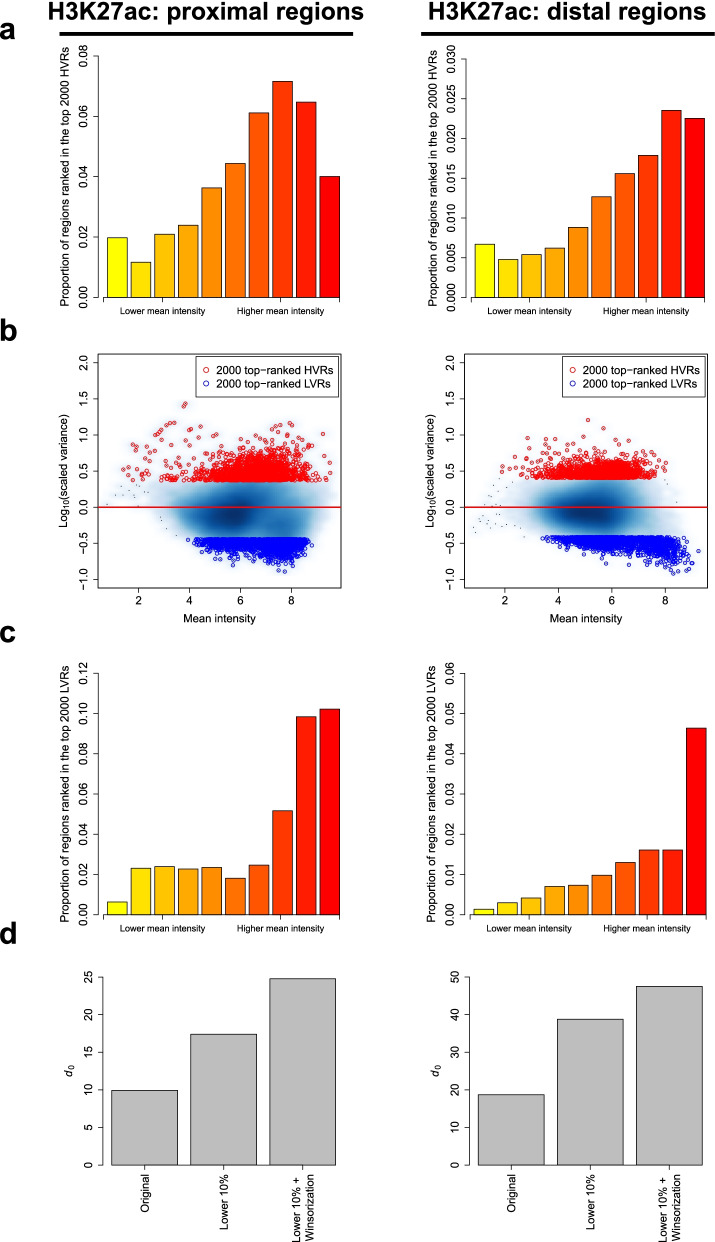
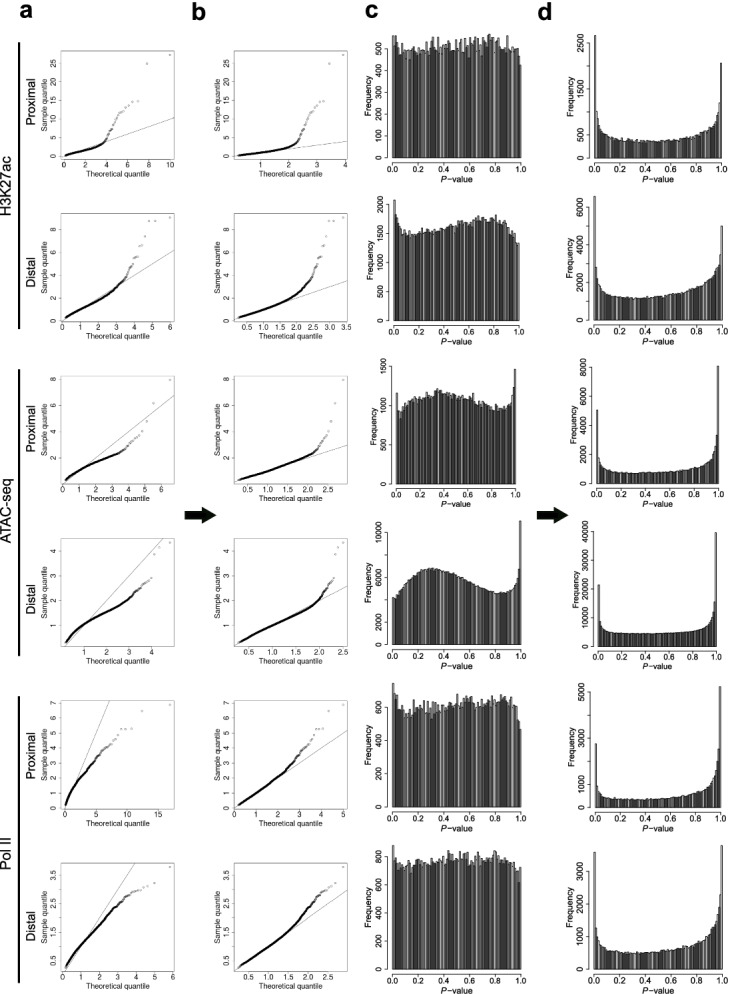
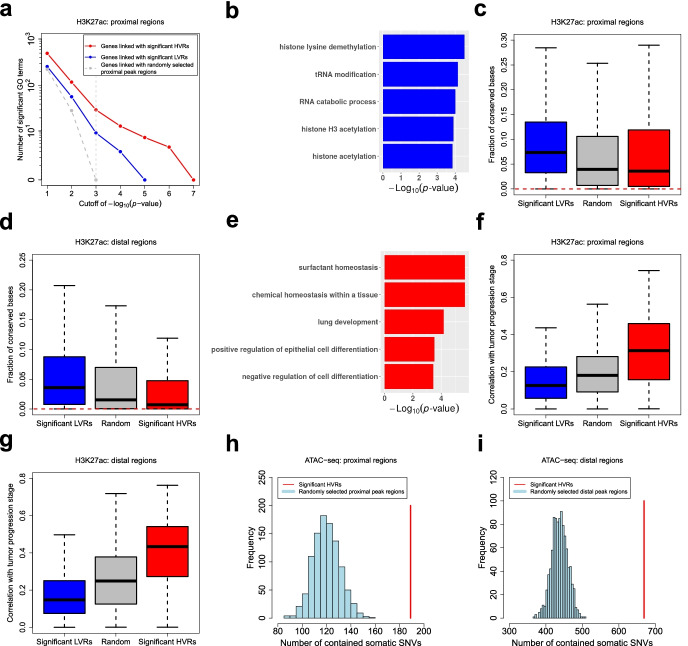
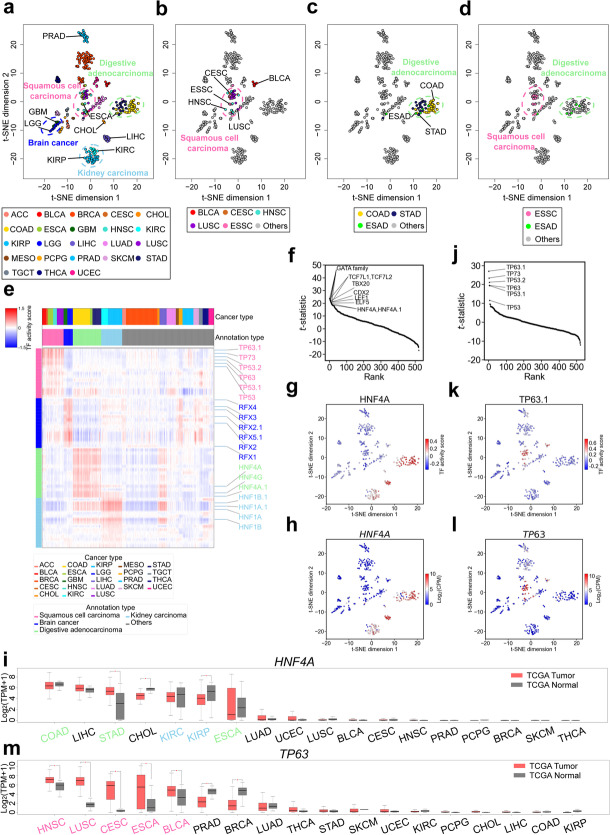
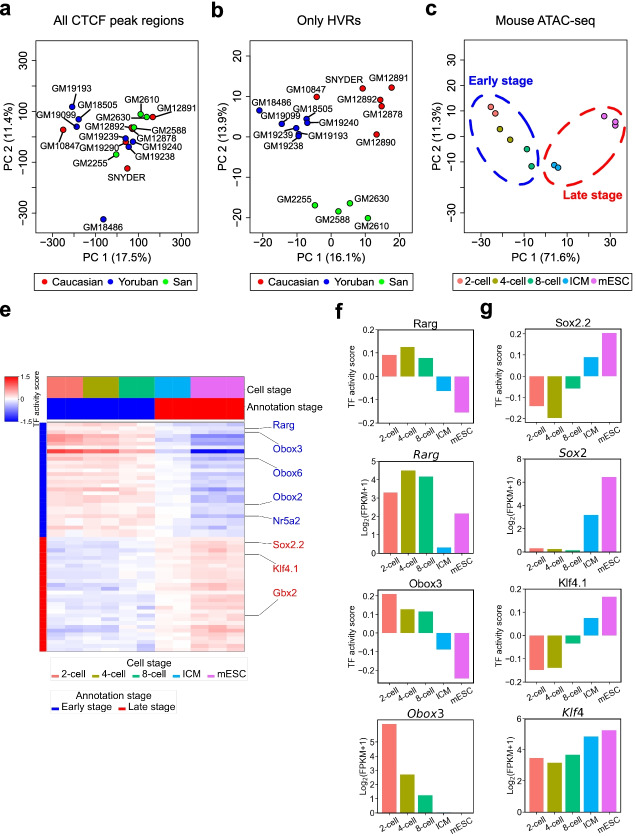
Identifying genomic regions with hypervariable ChIP-seq or ATAC-seq signals across given samples is essential for large-scale epigenetic studies. In particular, the hypervariable regions across tumors from different patients indicate their heterogeneity and can contribute to revealing potential cancer subtypes and the associated epigenetic markers. We present HyperChIP as the first complete statistical tool for the task. HyperChIP uses scaled variances that account for the mean-variance dependence to rank genomic regions, and it increases the statistical power by diminishing the influence of true hypervariable regions on model fitting. A pan-cancer case study illustrates the practical utility of HyperChIP.
识别给定样本中具有高可变 ChIP-seq 或 ATAC-seq 信号的基因组区域对于大规模表观遗传学研究至关重要。特别是,来自不同患者的肿瘤之间的高可变区域表明了它们的异质性,并有助于揭示潜在的癌症亚型和相关的表观遗传标记。我们提出了 HyperChIP,这是第一个用于该任务的完整统计工具。HyperChIP 使用缩放方差来考虑均值-方差依赖性对基因组区域进行排名,并通过减少真实高可变区域对模型拟合的影响来提高统计能力。一个泛癌症案例研究说明了 HyperChIP 的实际效用。