Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA 98195, USA.
Department of Human Genetics, Radboud University Medical Center, 6500 Nijmegen, the Netherlands; Laboratory of Cancer Genetics and Tumor Biology, Cancer and Translational Medicine Research Unit and Biocenter Oulu, University of Oulu, 90220 Oulu, Finland.
Am J Hum Genet. 2022 Apr 7;109(4):631-646. doi: 10.1016/j.ajhg.2022.02.014. Epub 2022 Mar 14.
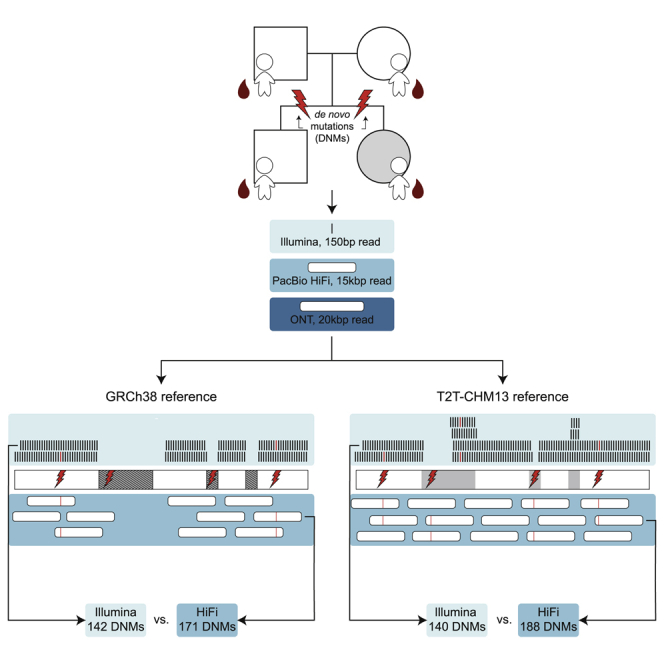
Studies of de novo mutation (DNM) have typically excluded some of the most repetitive and complex regions of the genome because these regions cannot be unambiguously mapped with short-read sequencing data. To better understand the genome-wide pattern of DNM, we generated long-read sequence data from an autism parent-child quad with an affected female where no pathogenic variant had been discovered in short-read Illumina sequence data. We deeply sequenced all four individuals by using three sequencing platforms (Illumina, Oxford Nanopore, and Pacific Biosciences) and three complementary technologies (Strand-seq, optical mapping, and 10X Genomics). Using long-read sequencing, we initially discovered and validated 171 DNMs across two children-a 20% increase in the number of de novo single-nucleotide variants (SNVs) and indels when compared to short-read callsets. The number of DNMs further increased by 5% when considering a more complete human reference (T2T-CHM13) because of the recovery of events in regions absent from GRCh38 (e.g., three DNMs in heterochromatic satellites). In total, we validated 195 de novo germline mutations and 23 potential post-zygotic mosaic mutations across both children; the overall true substitution rate based on this integrated callset is at least 1.41 × 10 substitutions per nucleotide per generation. We also identified six de novo insertions and deletions in tandem repeats, two of which represent structural variants. We demonstrate that long-read sequencing and assembly, especially when combined with a more complete reference genome, increases the number of DNMs by >25% compared to previous studies, providing a more complete catalog of DNM compared to short-read data alone.
研究从头突变 (DNM) 通常排除了基因组中最重复和最复杂的区域,因为这些区域无法用短读测序数据进行明确映射。为了更好地了解 DNM 的全基因组模式,我们从一名患有自闭症的女性的亲子四联体中生成了长读序列数据,在短读 Illumina 序列数据中未发现致病变体。我们使用三种测序平台(Illumina、Oxford Nanopore 和 Pacific Biosciences)和三种互补技术(Strand-seq、光学图谱和 10X Genomics)对所有四个人进行了深度测序。使用长读测序,我们最初在两个孩子中发现并验证了 171 个 DNM——与短读测序结果相比,新发现的单核苷酸变异 (SNV) 和插入缺失的数量增加了 20%。当考虑到一个更完整的人类参考(T2T-CHM13)时,DNM 的数量进一步增加了 5%,因为在 GRCh38 中不存在的区域(例如,异染色质卫星中的三个 DNM)中恢复了事件。总共,我们在两个孩子中验证了 195 个新的生殖系突变和 23 个潜在的合子后镶嵌突变;基于这个综合数据集的总体真实替换率至少为每代每个核苷酸 1.41×10 次替换。我们还在串联重复中鉴定了六个新的插入和缺失,其中两个代表结构变体。我们证明,长读测序和组装,尤其是与更完整的参考基因组结合使用时,与之前的研究相比,DNM 的数量增加了超过 25%,与单独使用短读数据相比,提供了更完整的 DNM 目录。