Ministry of Education Key Laboratory of Bioinformatics; Bioinformatics Division, Beijing National Research Center for Information Science and Technology; Center for Synthetic and Systems Biology, Department of Automation, Tsinghua University, Beijing 100084, China; Department of Statistics, Stanford University, Stanford, CA 94305, USA.
Ministry of Education Key Laboratory of Bioinformatics; Bioinformatics Division, Beijing National Research Center for Information Science and Technology; Center for Synthetic and Systems Biology, Department of Automation, Tsinghua University, Beijing 100084, China.
Genomics Proteomics Bioinformatics. 2022 Jun;20(3):496-507. doi: 10.1016/j.gpb.2021.08.015. Epub 2022 Mar 12.
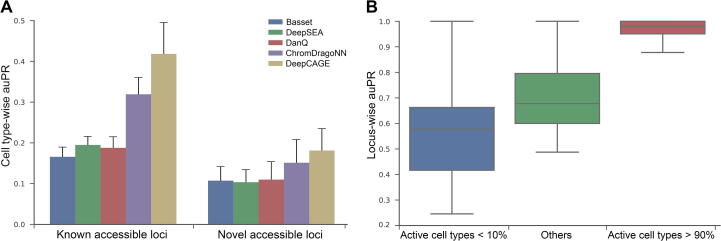
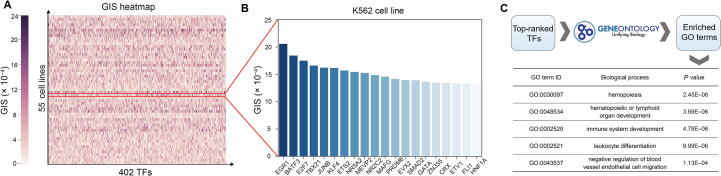
Although computational approaches have been complementing high-throughput biological experiments for the identification of functional regions in the human genome, it remains a great challenge to systematically decipher interactions between transcription factors (TFs) and regulatory elements to achieve interpretable annotations of chromatin accessibility across diverse cellular contexts. To solve this problem, we propose DeepCAGE, a deep learning framework that integrates sequence information and binding statuses of TFs, for the accurate prediction of chromatin accessible regions at a genome-wide scale in a variety of cell types. DeepCAGE takes advantage of a densely connected deep convolutional neural network architecture to automatically learn sequence signatures of known chromatin accessible regions and then incorporates such features with expression levels and binding activities of human core TFs to predict novel chromatin accessible regions. In a series of systematic comparisons with existing methods, DeepCAGE exhibits superior performance in not only the classification but also the regression of chromatin accessibility signals. In a detailed analysis of TF activities, DeepCAGE successfully extracts novel binding motifs and measures the contribution of a TF to the regulation with respect to a specific locus in a certain cell type. When applied to whole-genome sequencing data analysis, our method successfully prioritizes putative deleterious variants underlying a human complex trait and thus provides insights into the understanding of disease-associated genetic variants. DeepCAGE can be downloaded from https://github.com/kimmo1019/DeepCAGE.
虽然计算方法一直在为识别人类基因组中的功能区域补充高通量生物学实验,但系统破译转录因子 (TF) 和调控元件之间的相互作用,以实现对不同细胞环境下染色质可及性的可解释注释,仍然是一个巨大的挑战。为了解决这个问题,我们提出了 DeepCAGE,这是一个深度学习框架,它整合了序列信息和 TF 的结合状态,用于在多种细胞类型中准确预测全基因组范围内的染色质可及区域。DeepCAGE 利用密集连接的深度卷积神经网络架构,自动学习已知染色质可及区域的序列特征,然后将这些特征与人类核心 TF 的表达水平和结合活性相结合,以预测新的染色质可及区域。在与现有方法的一系列系统比较中,DeepCAGE 在分类和回归染色质可及性信号方面都表现出了优异的性能。在对 TF 活性的详细分析中,DeepCAGE 成功提取了新的结合基序,并测量了 TF 相对于特定细胞类型中特定基因座的调控贡献。当应用于全基因组测序数据分析时,我们的方法成功地确定了人类复杂性状潜在的有害变异,从而深入了解与疾病相关的遗传变异。DeepCAGE 可从 https://github.com/kimmo1019/DeepCAGE 下载。