Center for Neuroscience Research, Children's National Research Institute, Children's National Hospital, 111 Michigan Avenue, Washington, DC, 20010, USA.
Department of Pediatrics, Division of Neonatology, Children's National Hospital, Washington DC, USA.
J Neurodev Disord. 2022 Mar 29;14(1):26. doi: 10.1186/s11689-022-09431-3.
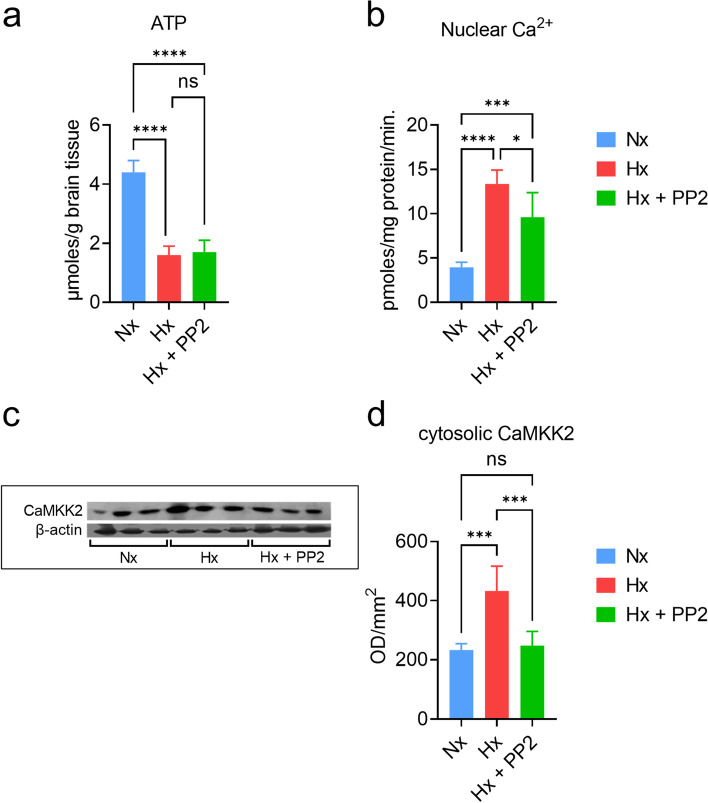
Neonatal hypoxic brain injury is a major cause of intellectual and developmental disability. Hypoxia causes neuronal dysfunction and death in the developing cerebral cortex due to excitotoxic Ca-influx. In the translational piglet model of hypoxic encephalopathy, we have previously shown that hypoxia overactivates Ca/Calmodulin (CaM) signaling via Sarcoma (Src) kinase in cortical neurons, resulting in overexpression of proapoptotic genes. However, identifying the exact relationship between alterations in neuronal Ca-influx, molecular determinants of cell death, and the degree of hypoxia in a dynamic system represents a significant challenge.
We used experimental and computational methods to identify molecular events critical to the onset of excitotoxicity-induced apoptosis in the cerebral cortex of newborn piglets. We used 2-3-day-old piglets (normoxic [Nx], hypoxic [Hx], and hypoxic + Src-inhibitor-treatment [Hx+PP2] groups) for biochemical analysis of ATP production, Ca-influx, and Ca/CaM-dependent protein kinase kinase 2 (CaMKK2) expression. We then used SimBiology to build a computational model of the Ca/CaM-Src-kinase signaling cascade, simulating Nx, Hx, and Hx+PP2 conditions. To evaluate our model, we used Sobol variance decomposition, multiparametric global sensitivity analysis, and parameter scanning.
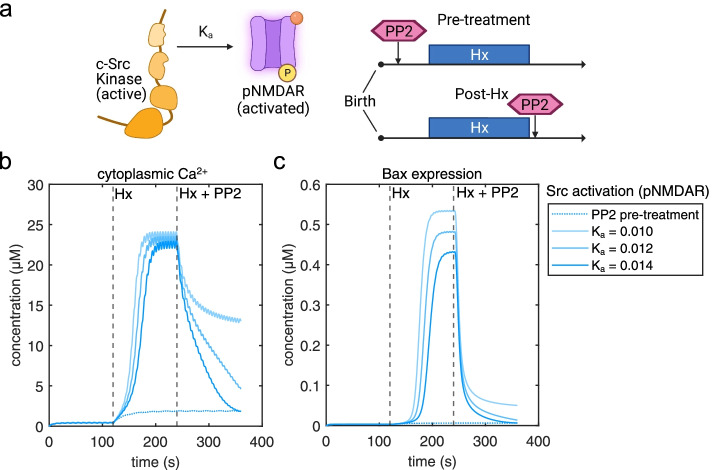
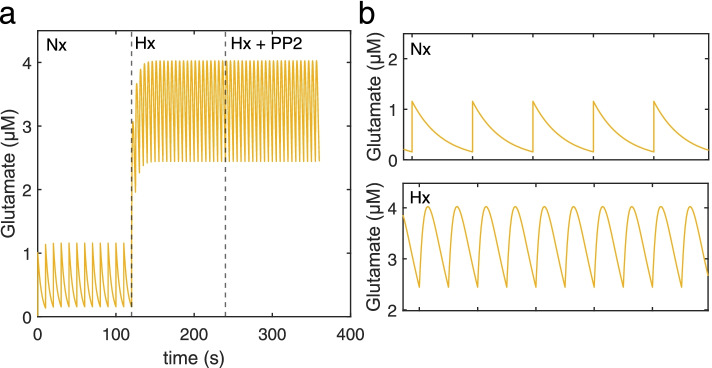
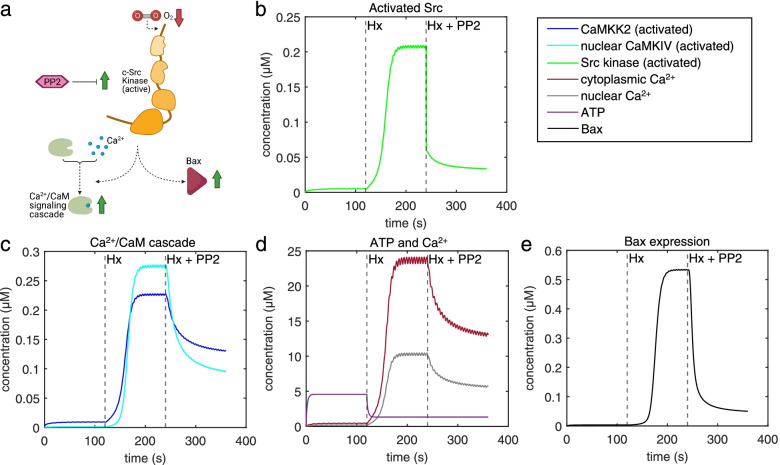
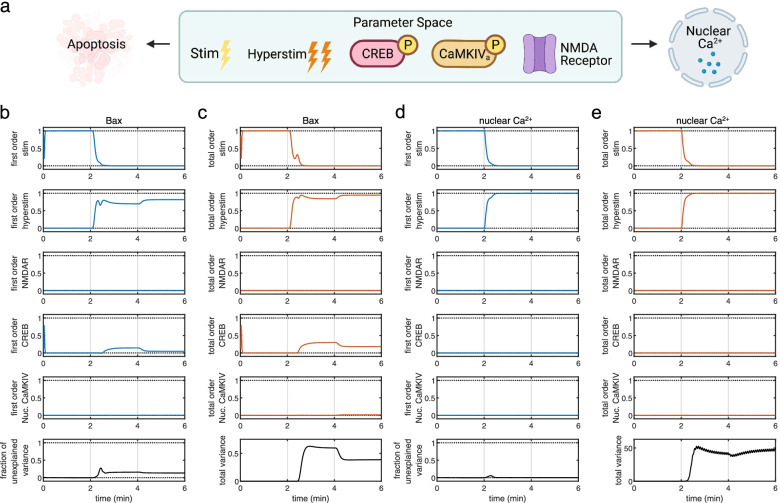
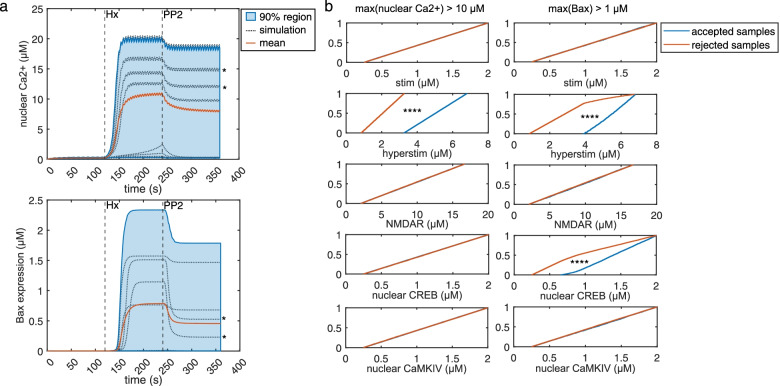
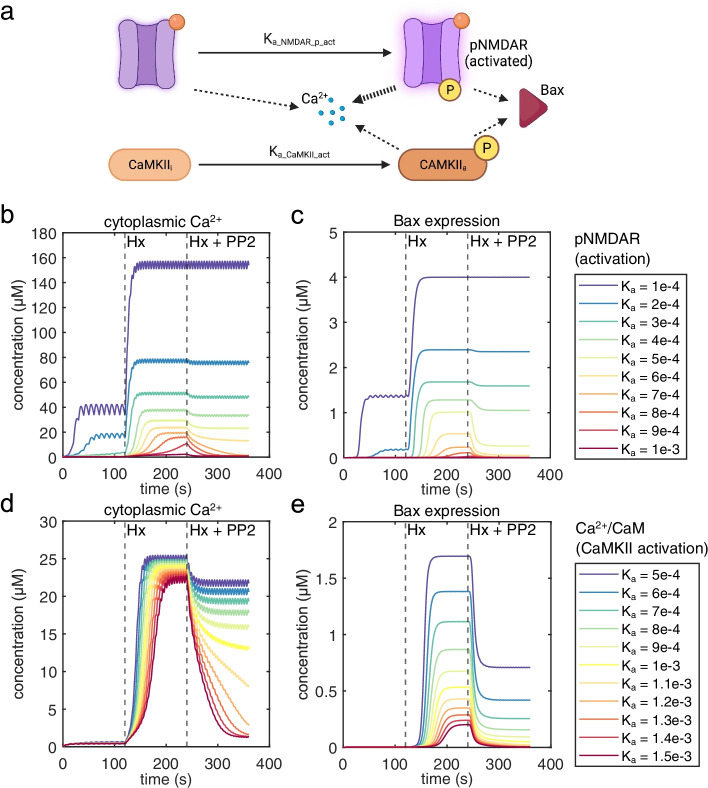
Our model captures important molecular trends caused by hypoxia in the piglet brain. Incorporating the action of Src kinase inhibitor PP2 further validated our model and enabled predictive analysis of the effect of hypoxia on CaMKK2. We determined the impact of a feedback loop related to Src phosphorylation of NMDA receptors and activation kinetics of CaMKII. We also identified distinct modes of signaling wherein Ca level alterations following Src kinase inhibition may not be a linear predictor of changes in Bax expression. Importantly, our model indicates that while pharmacological pre-treatment significantly reduces the onset of abnormal Ca-influx, there exists a window of intervention after hypoxia during which targeted modulation of Src-NMDAR interaction kinetics in combination with PP2 administration can reduce Ca-influx and Bax expression to similar levels as pre-treatment.
Our model identifies new dynamics of critical components in the Ca/CaM-Src signaling pathway leading to neuronal injury and provides a feasible framework for drug efficacy studies in translational models of neonatal brain injury for the prevention of intellectual and developmental disabilities.
新生儿缺氧性脑损伤是智力和发育障碍的主要原因。缺氧会导致发育中的大脑皮层神经元功能障碍和死亡,这是由于兴奋毒性 Ca 流入引起的。在缺氧脑病的转化仔猪模型中,我们之前已经表明,缺氧通过肉瘤(Src)激酶使皮质神经元中的 Ca/钙调蛋白(CaM)信号过度激活,导致促凋亡基因的过度表达。然而,在动态系统中确定神经元 Ca 流入、细胞死亡的分子决定因素和缺氧程度之间的确切关系是一项重大挑战。
我们使用实验和计算方法来确定对新生仔猪大脑皮质兴奋毒性诱导细胞凋亡起始至关重要的分子事件。我们使用 2-3 天大的仔猪(正常氧合 [Nx]、缺氧 [Hx]和缺氧+Src 抑制剂治疗 [Hx+PP2]组)进行生化分析,以分析 ATP 产生、Ca 流入和 Ca/CaM 依赖性蛋白激酶激酶 2(CaMKK2)表达。然后,我们使用 SimBiology 构建 Ca/CaM-Src 激酶信号级联的计算模型,模拟 Nx、Hx 和 Hx+PP2 条件。为了评估我们的模型,我们使用 Sobol 方差分解、多参数全局敏感性分析和参数扫描。
我们的模型捕捉到了仔猪大脑缺氧引起的重要分子趋势。加入 Src 激酶抑制剂 PP2 进一步验证了我们的模型,并能够预测缺氧对 CaMKK2 的影响。我们确定了与 Src 磷酸化 NMDA 受体和 CaMKII 激活动力学相关的反馈环的影响。我们还发现了不同的信号模式,其中 Src 激酶抑制后 Ca 水平的改变可能不是 Bax 表达变化的线性预测因子。重要的是,我们的模型表明,虽然药物预处理显著降低了异常 Ca 流入的发生,但在缺氧后存在一个干预窗口,在此期间,靶向调节 Src-NMDAR 相互作用动力学并结合 PP2 给药可以将 Ca 流入和 Bax 表达降低到与预处理相似的水平。
我们的模型确定了导致神经元损伤的 Ca/CaM-Src 信号通路中关键成分的新动态,并为新生儿脑损伤转化模型中的药物疗效研究提供了可行的框架,以预防智力和发育障碍。