Department of Laboratory Medicine and Pathology, University of Washington School of Medicine, Seattle, WA, 98102, USA.
Vaccine and Infectious Disease Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA.
Sci Rep. 2022 Apr 7;12(1):5856. doi: 10.1038/s41598-022-09752-2.
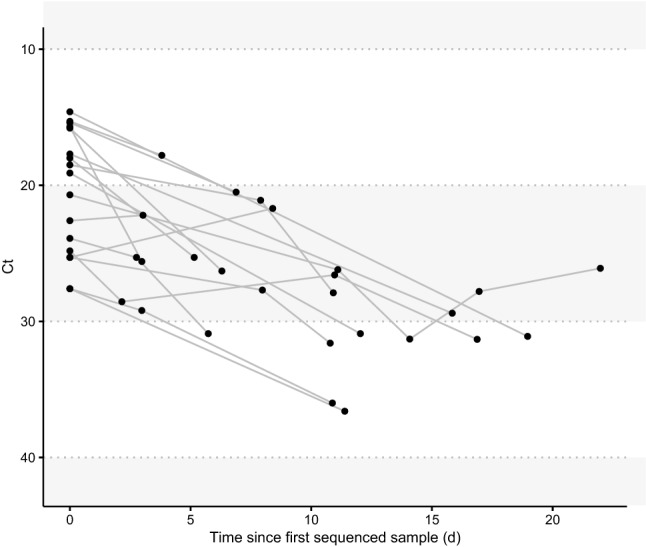
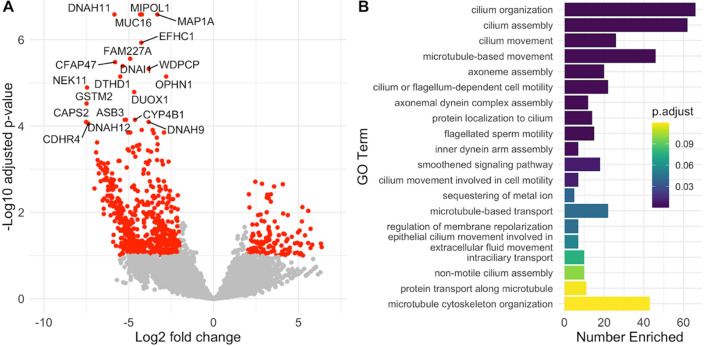
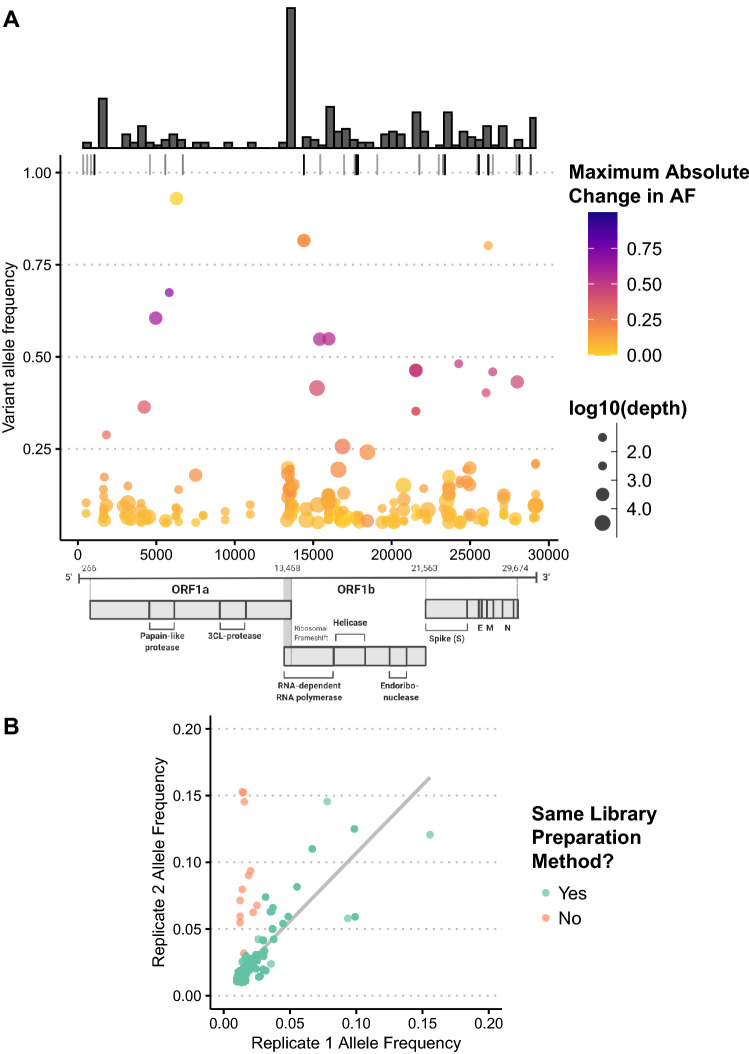
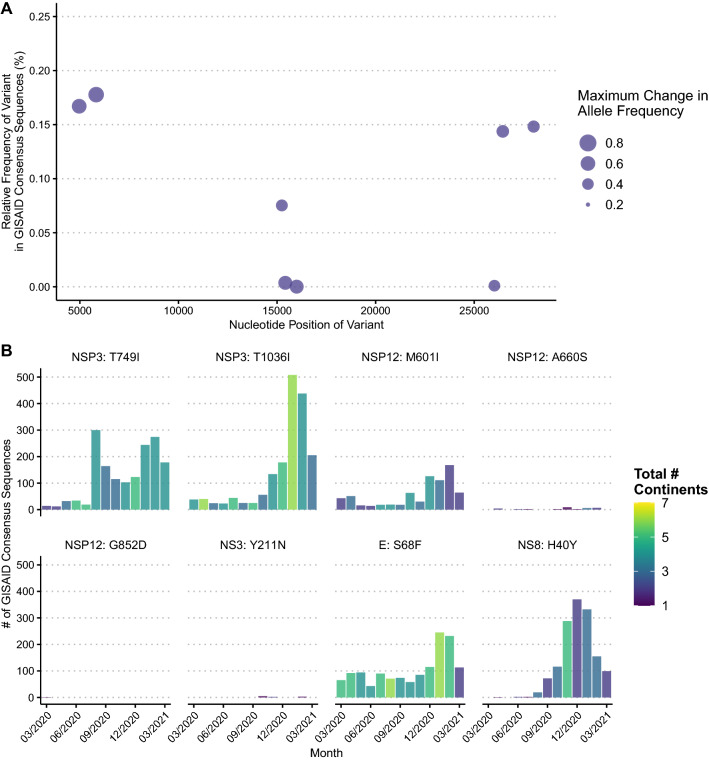
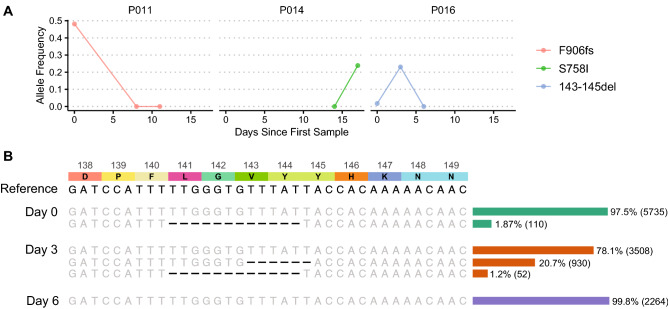
Rapid dissemination of SARS-CoV-2 sequencing data to public repositories has enabled widespread study of viral genomes, but studies of longitudinal specimens from infected persons are relatively limited. Analysis of longitudinal specimens enables understanding of how host immune pressures drive viral evolution in vivo. Here we performed sequencing of 49 longitudinal SARS-CoV-2-positive samples from 20 patients in Washington State collected between March and September of 2020. Viral loads declined over time with an average increase in RT-QPCR cycle threshold of 0.87 per day. We found that there was negligible change in SARS-CoV-2 consensus sequences over time, but identified a number of nonsynonymous variants at low frequencies across the genome. We observed enrichment for a relatively small number of these variants, all of which are now seen in consensus genomes across the globe at low prevalence. In one patient, we saw rapid emergence of various low-level deletion variants at the N-terminal domain of the spike glycoprotein, some of which have previously been shown to be associated with reduced neutralization potency from sera. In a subset of samples that were sequenced using metagenomic methods, differential gene expression analysis showed a downregulation of cytoskeletal genes that was consistent with a loss of ciliated epithelium during infection and recovery. We also identified co-occurrence of bacterial species in samples from multiple hospitalized individuals. These results demonstrate that the intrahost genetic composition of SARS-CoV-2 is dynamic during the course of COVID-19, and highlight the need for continued surveillance and deep sequencing of minor variants.
SARS-CoV-2 测序数据迅速传播到公共存储库,使人们能够广泛研究病毒基因组,但对感染患者的纵向样本的研究相对有限。对纵向样本的分析使人们能够了解宿主免疫压力如何在体内驱动病毒进化。在这里,我们对 2020 年 3 月至 9 月期间在华盛顿州采集的 20 名患者的 49 份 SARS-CoV-2 阳性纵向样本进行了测序。病毒载量随时间下降,平均每天 RT-QPCR 循环阈值增加 0.87。我们发现,SARS-CoV-2 共识序列随时间几乎没有变化,但在整个基因组中发现了许多低频的非同义变体。我们观察到这些变体中有相当数量的富集,所有这些变体现在在全球共识基因组中都以低流行率出现。在一名患者中,我们观察到 Spike 糖蛋白 N 端结构域的各种低水平缺失变体迅速出现,其中一些先前已被证明与血清中和效力降低有关。在使用宏基因组方法测序的一部分样本中,差异基因表达分析显示细胞骨架基因下调,这与感染和恢复过程中纤毛上皮细胞丢失一致。我们还在多个住院患者的样本中鉴定出细菌物种的共同出现。这些结果表明,在 COVID-19 期间,SARS-CoV-2 的宿主内遗传组成是动态的,并强调需要对次要变体进行持续监测和深度测序。