Perera Duwage C, Rasaiah Jayendran C
Department of Chemistry, University of Maine, Orono, Maine 04469, United States.
ACS Omega. 2022 Apr 4;7(15):12556-12569. doi: 10.1021/acsomega.1c05666. eCollection 2022 Apr 19.
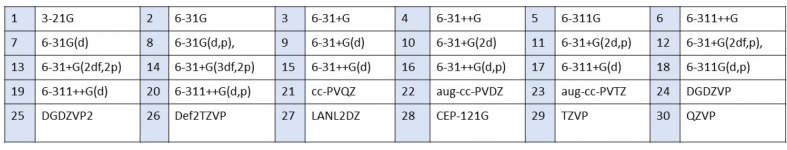
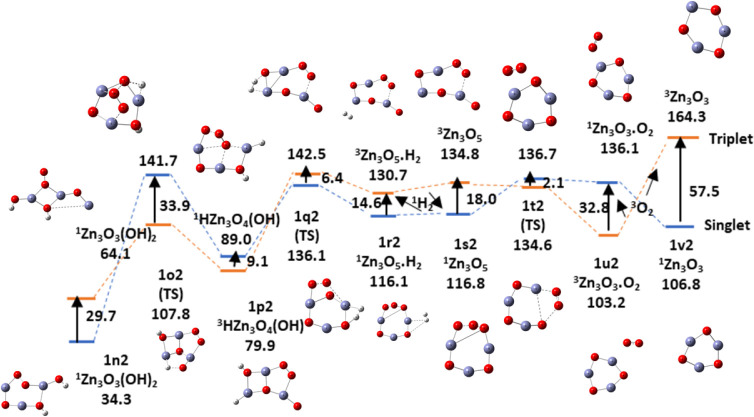
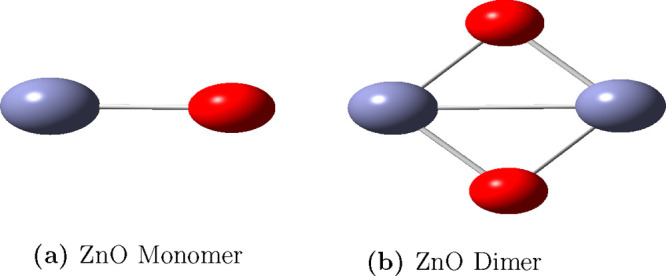
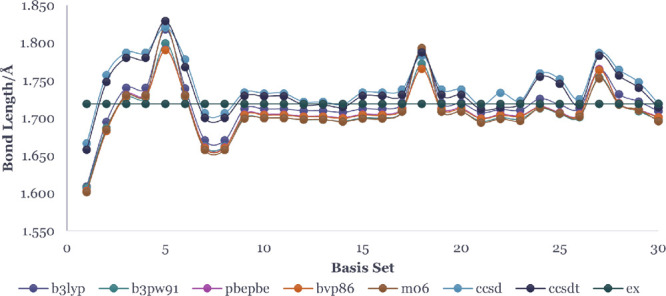
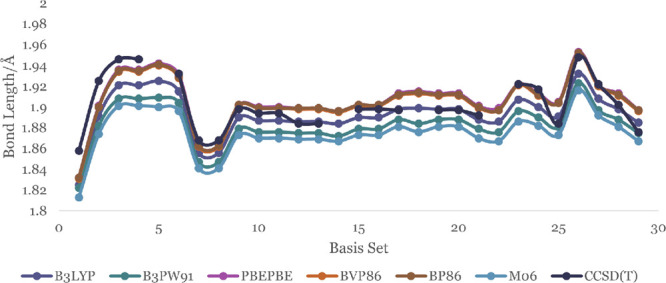
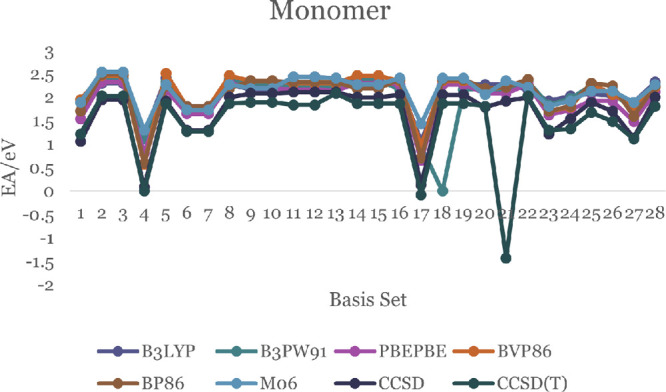
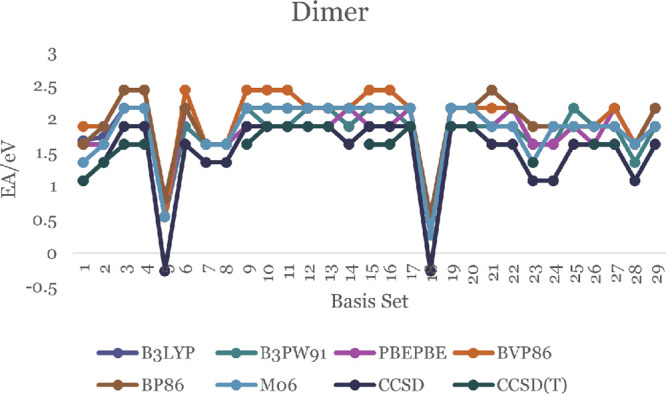
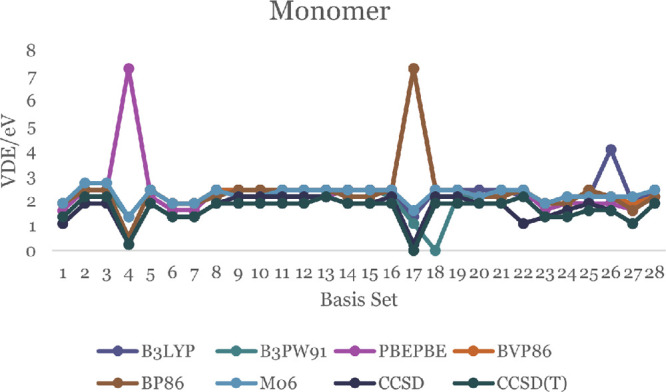
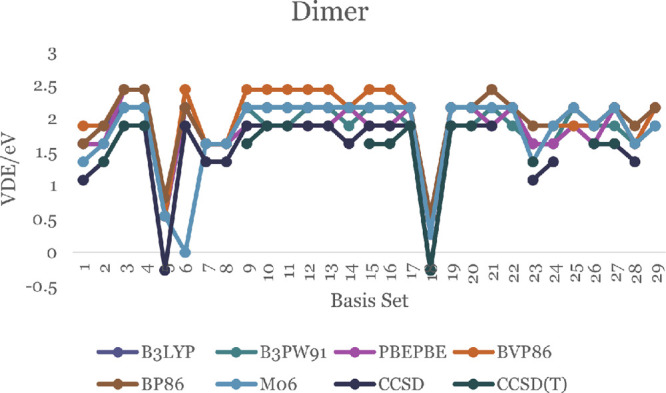
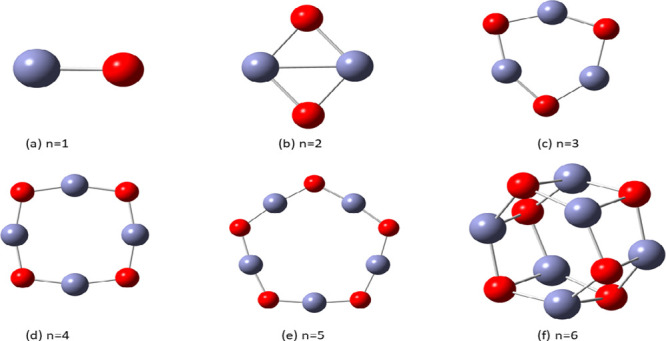
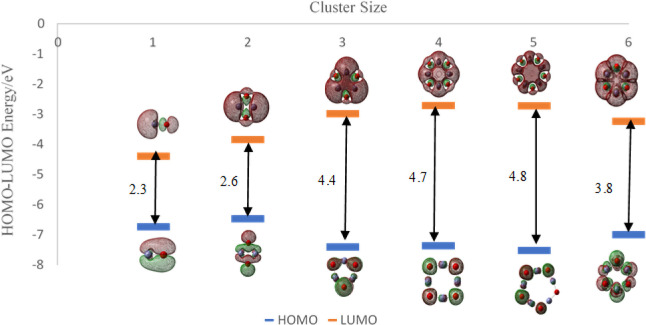
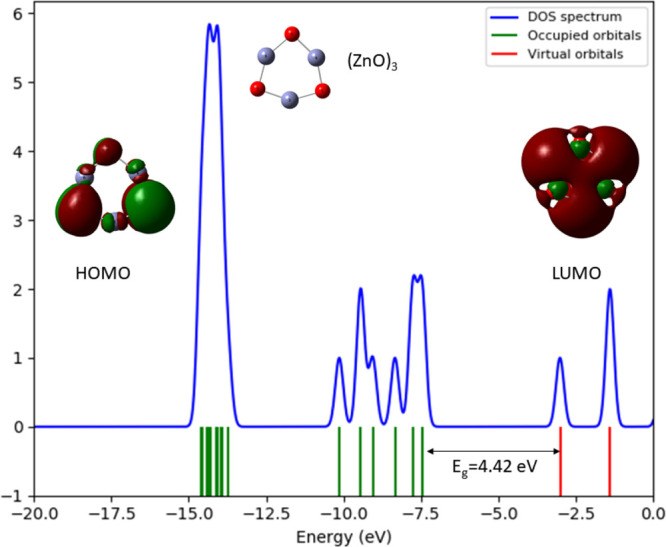
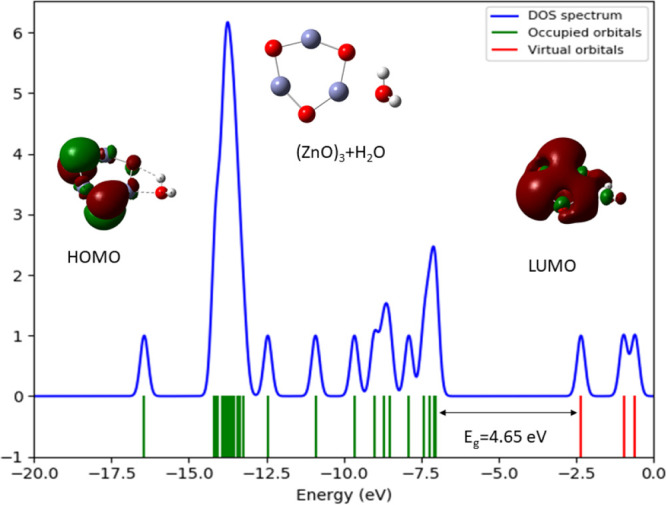
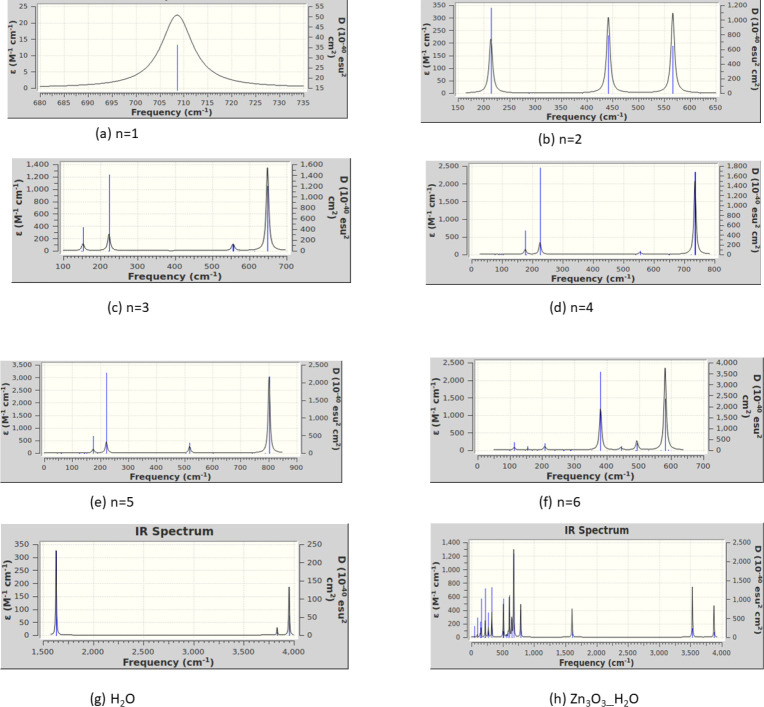
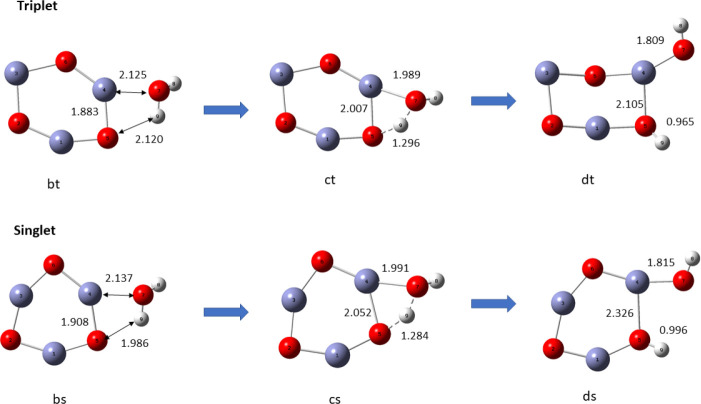
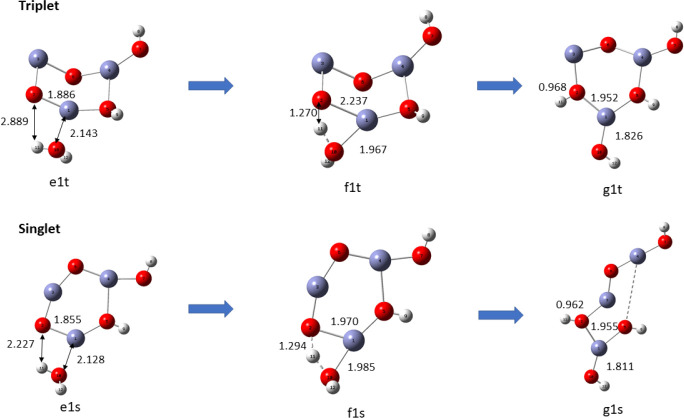
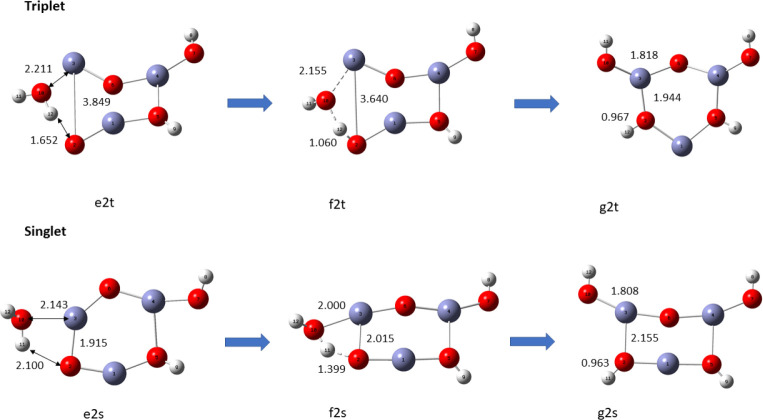
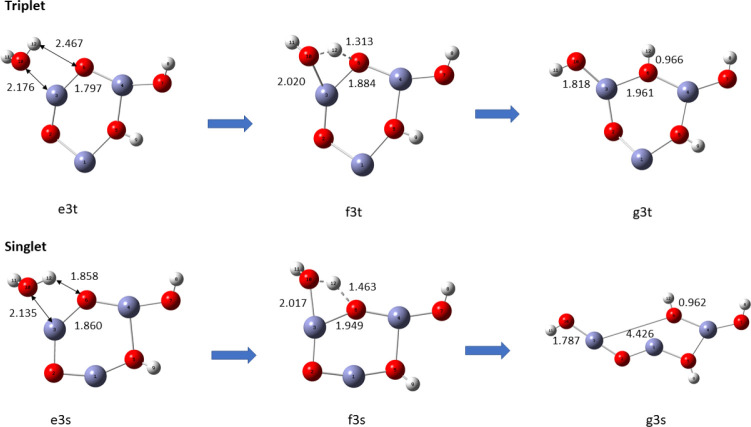
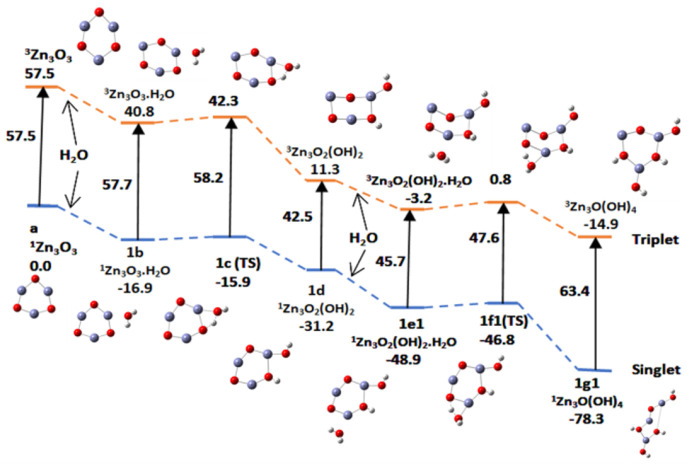
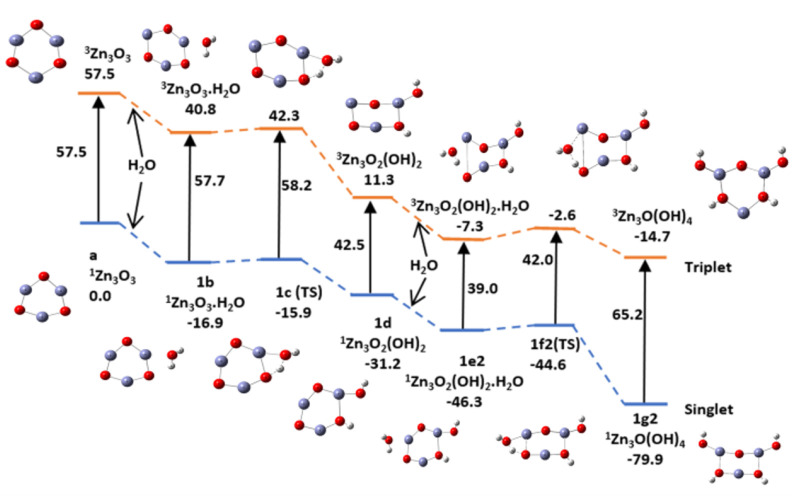
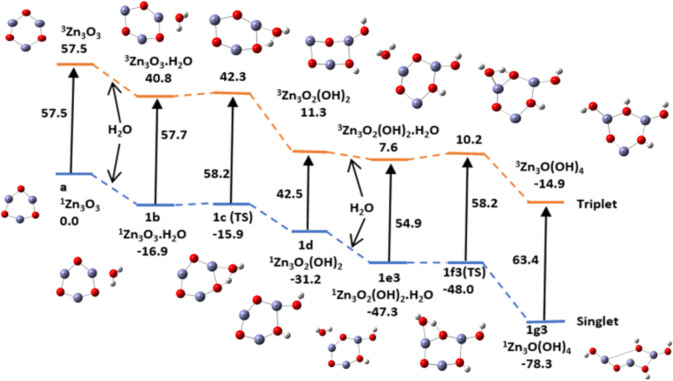
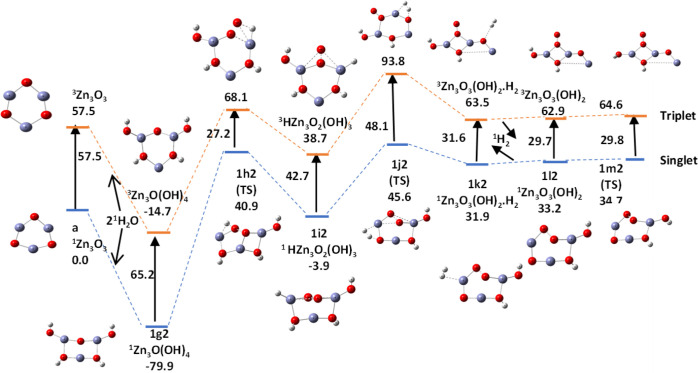
In this communication, we use density functional theory (DFT) to study the structural (geometry) and electronic properties (vertical detachment energy and electron affinity) of ZnO monomers and dimers that can be used to form ZnO clusters of different sizes, with a view to adapting one or more of them as catalysts or photocatalysts, standing alone or on suitable substrates like graphene, to split water. We also investigate different pairs of exchange functionals and basis sets to optimize their choice in our DFT calculations and to compare the singlet-triplet energy gaps of small ZnO clusters of different sizes to select an optimal cluster size for water splitting. We find that the B3LYP/DGDZVP2 exchange functional/basis set is a reliable combination for use with DFT to calculate the geometry and electronic properties of small ZnO nanoclusters from among several other combinations of exchange functionals and basis sets. Comparisons of the singlet-triplet energy gaps show that the trimer (ZnO) has an energy gap of 58.66 k cal/mol. which is approximately equal to the energy of a visible photon at a wavelength of 500 nm, and a HOMO-LUMO gap of 4.4 eV, making it a suitable choice of photocatalyst for the oxidation of water from among six (ZnO) nanoclusters of monomers, with ranging from 1 to 6. We used this exchange functional/basis set to study the structural and energetic details of hydration and hydrolysis of water absorbed on the (ZnO) nanocatalyst and calculated the corresponding potential energy profiles to identify three sets of singlet-triplet pathways for water splitting. Detailed study of a pathway showed that oxygen is produced after hydrogen, and the rate-determining step is the formation of hydrogen.
在本通讯中,我们使用密度泛函理论(DFT)来研究可用于形成不同尺寸ZnO团簇的ZnO单体和二聚体的结构(几何形状)和电子性质(垂直脱附能和电子亲和势),旨在将其中一种或多种用作单独或负载于石墨烯等合适基底上的催化剂或光催化剂来分解水。我们还研究了不同的交换泛函和基组对,以优化其在DFT计算中的选择,并比较不同尺寸的小ZnO团簇的单重态 - 三重态能隙,以选择用于水分解的最佳团簇尺寸。我们发现,在交换泛函和基组的其他几种组合中,B3LYP/DGDZVP2交换泛函/基组是与DFT结合使用以计算小ZnO纳米团簇几何形状和电子性质的可靠组合。单重态 - 三重态能隙的比较表明,三聚体(ZnO)的能隙为58.66千卡/摩尔,这大约等于波长为500nm的可见光子的能量,其最高占据分子轨道 - 最低未占据分子轨道(HOMO - LUMO)能隙为4.4eV,使其成为六种(ZnO)单体纳米团簇(范围从1到6)中用于水氧化的光催化剂的合适选择。我们使用此交换泛函/基组研究了吸附在(ZnO)纳米催化剂上的水的水合和水解的结构和能量细节,并计算了相应的势能曲线,以确定水分解的三组单重态 - 三重态途径。对一条途径的详细研究表明,氢之后产生氧,且速率决定步骤是氢的形成。