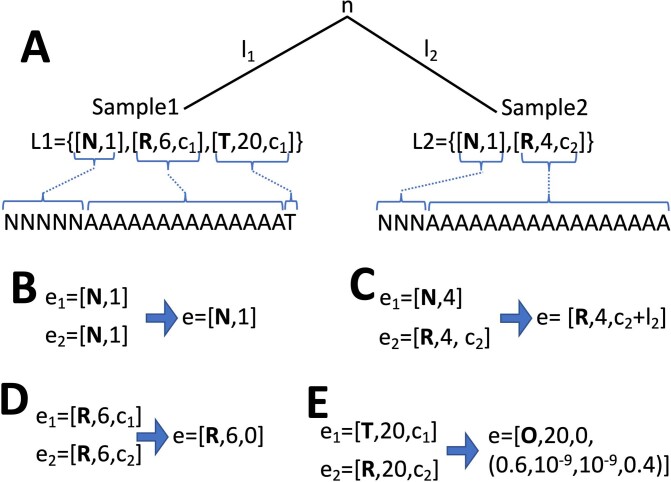
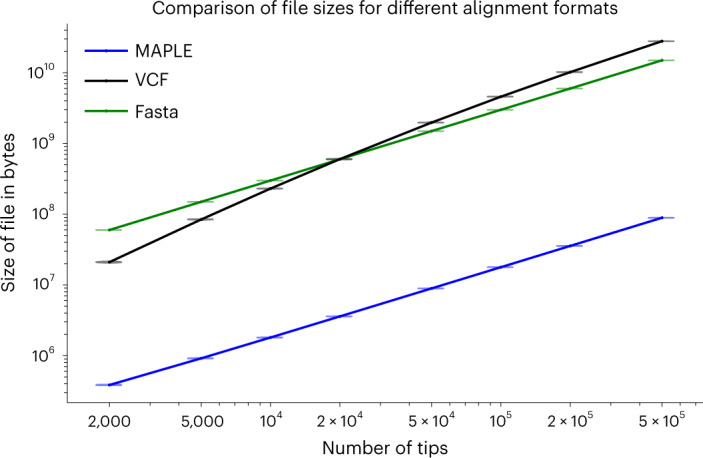
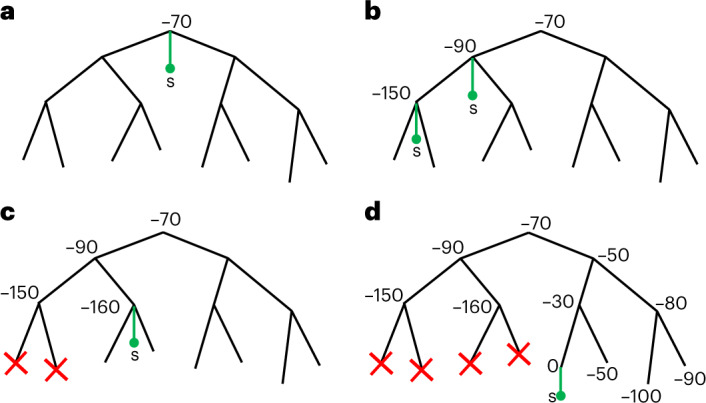
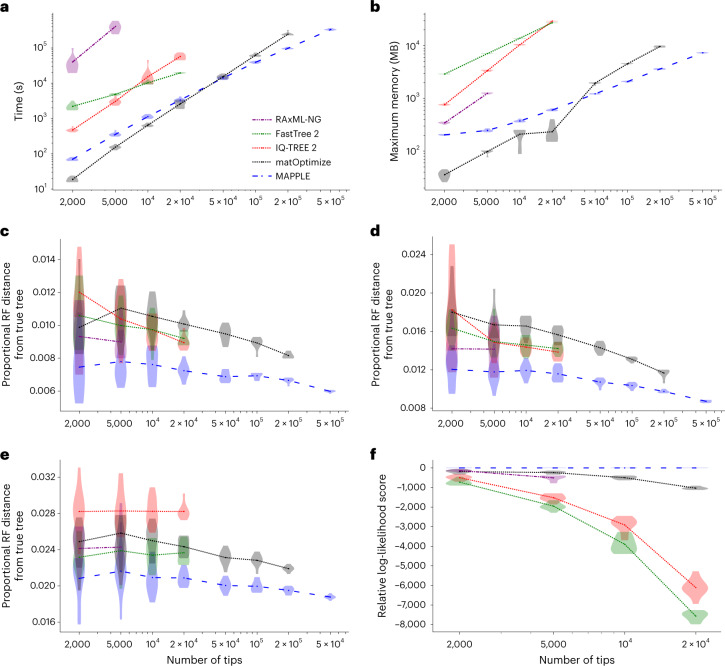
European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Hinxton, UK.
Max Planck Institute for Molecular Genetics, Berlin, Germany.
Nat Genet. 2023 May;55(5):746-752. doi: 10.1038/s41588-023-01368-0. Epub 2023 Apr 10.
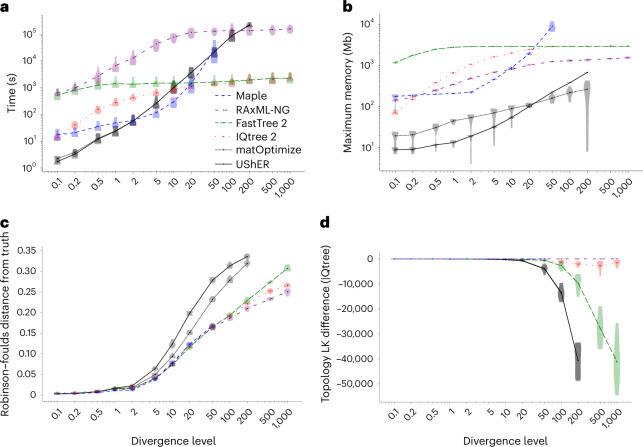
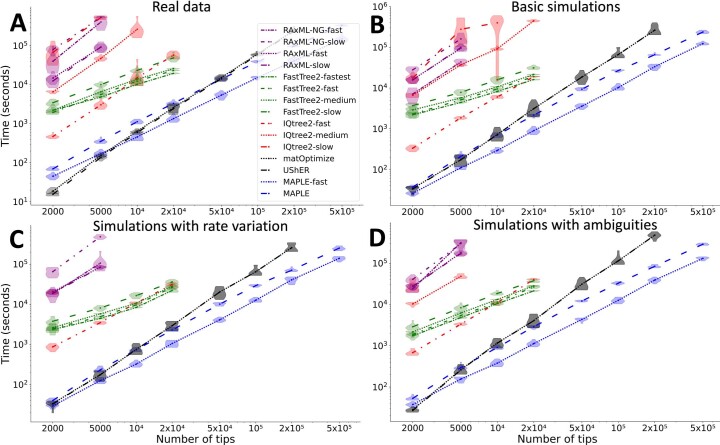
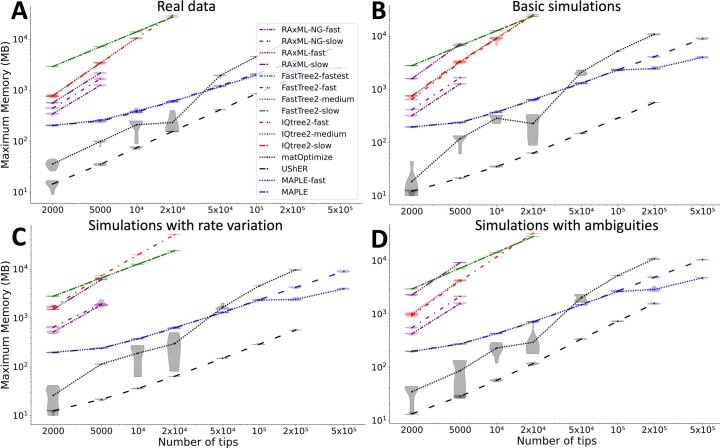
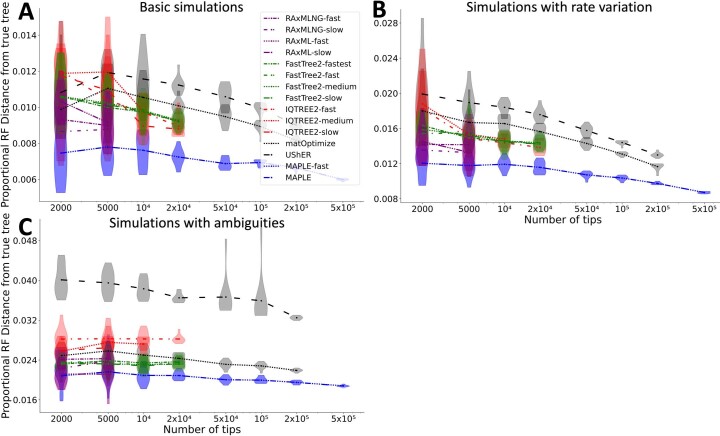
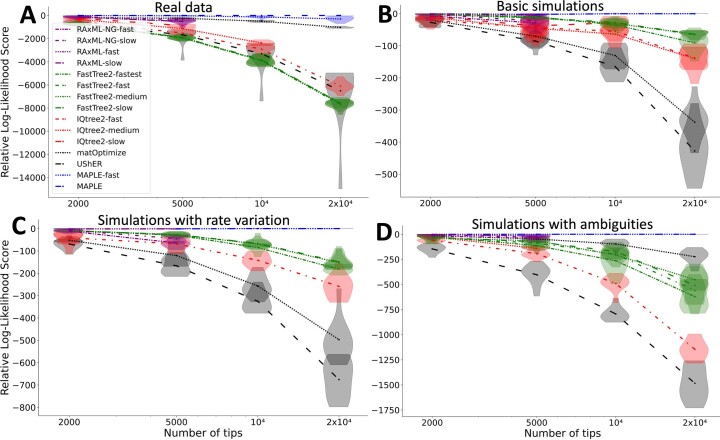
Phylogenetics has a crucial role in genomic epidemiology. Enabled by unparalleled volumes of genome sequence data generated to study and help contain the COVID-19 pandemic, phylogenetic analyses of SARS-CoV-2 genomes have shed light on the virus's origins, spread, and the emergence and reproductive success of new variants. However, most phylogenetic approaches, including maximum likelihood and Bayesian methods, cannot scale to the size of the datasets from the current pandemic. We present 'MAximum Parsimonious Likelihood Estimation' (MAPLE), an approach for likelihood-based phylogenetic analysis of epidemiological genomic datasets at unprecedented scales. MAPLE infers SARS-CoV-2 phylogenies more accurately than existing maximum likelihood approaches while running up to thousands of times faster, and requiring at least 100 times less memory on large datasets. This extends the reach of genomic epidemiology, allowing the continued use of accurate phylogenetic, phylogeographic and phylodynamic analyses on datasets of millions of genomes.
系统发生学在基因组流行病学中具有关键作用。得益于为研究和帮助控制 COVID-19 大流行而生成的无与伦比的大量基因组序列数据,对 SARS-CoV-2 基因组的系统发生分析揭示了该病毒的起源、传播以及新变体的出现和繁殖成功率。然而,包括最大似然法和贝叶斯方法在内的大多数系统发生方法都无法扩展到当前大流行数据集的规模。我们提出了“最大简约似然估计”(MAPLE),这是一种用于在前所未有的规模上对流行病学基因组数据集进行基于似然的系统发生分析的方法。MAPLE 比现有的最大似然方法更准确地推断 SARS-CoV-2 的系统发生关系,同时运行速度快数千倍,在大型数据集上所需的内存至少少 100 倍。这扩展了基因组流行病学的范围,允许在数百万个基因组数据集上继续使用准确的系统发生、系统地理学和系统动力学分析。