School of Computing, College of Engineering and Computer Science, Australian National University, Canberra, ACT 2600, Australia.
Ecology and Evolution, Research School of Biology, College of Science, Australian National University, Canberra, ACT 2600, Australia.
Mol Biol Evol. 2022 May 3;39(5). doi: 10.1093/molbev/msac092.
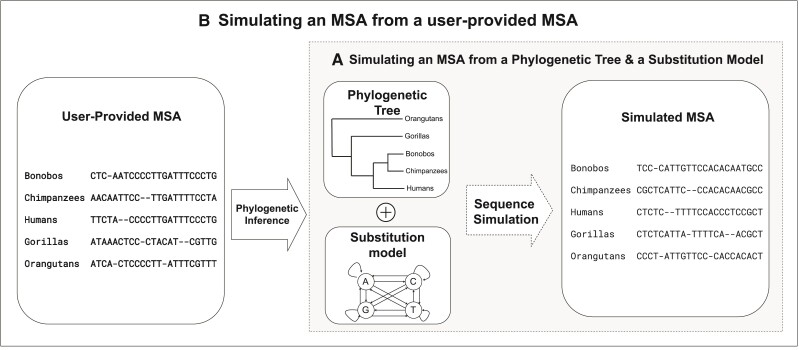
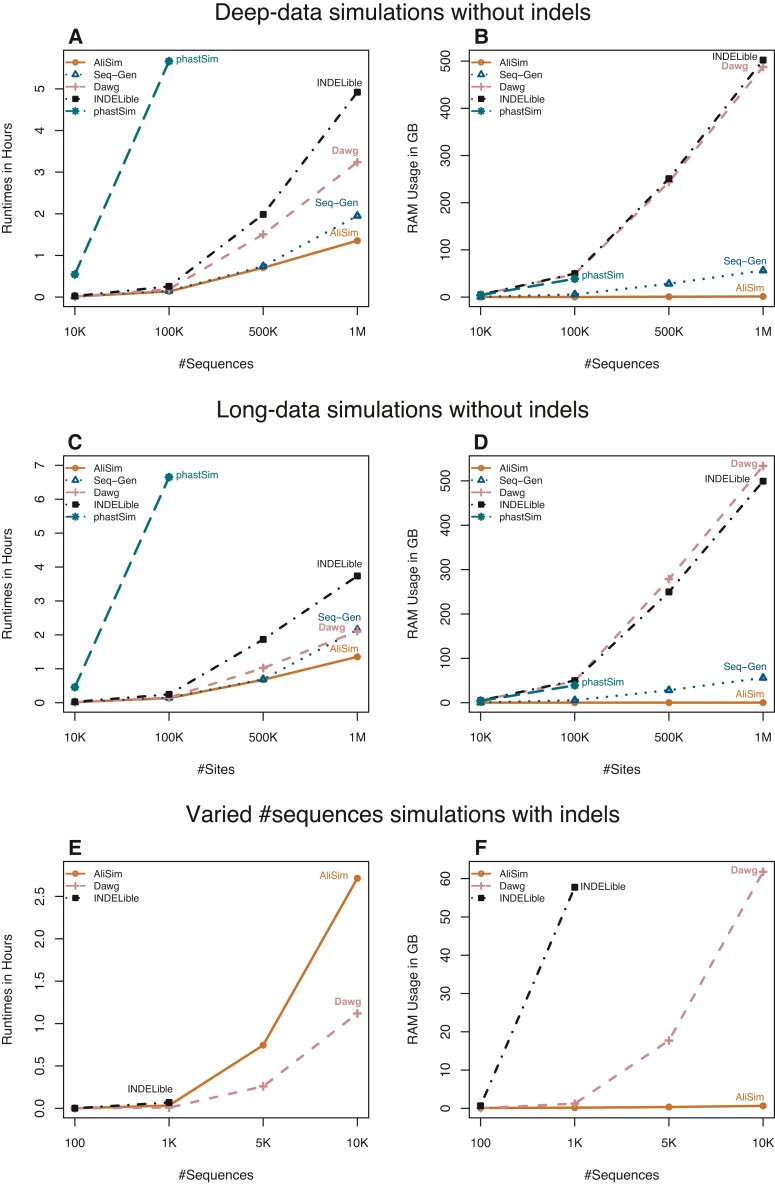
Sequence simulators play an important role in phylogenetics. Simulated data has many applications, such as evaluating the performance of different methods, hypothesis testing with parametric bootstraps, and, more recently, generating data for training machine-learning applications. Many sequence simulation programmes exist, but the most feature-rich programmes tend to be rather slow, and the fastest programmes tend to be feature-poor. Here, we introduce AliSim, a new tool that can efficiently simulate biologically realistic alignments under a large range of complex evolutionary models. To achieve high performance across a wide range of simulation conditions, AliSim implements an adaptive approach that combines the commonly used rate matrix and probability matrix approaches. AliSim takes 1.4 h and 1.3 GB RAM to simulate alignments with one million sequences or sites, whereas popular software Seq-Gen, Dawg, and INDELible require 2-5 h and 50-500 GB of RAM. We provide AliSim as an extension of the IQ-TREE software version 2.2, freely available at www.iqtree.org, and a comprehensive user tutorial at http://www.iqtree.org/doc/AliSim.
序列模拟器在系统发育学中起着重要作用。模拟数据有许多应用,例如评估不同方法的性能、使用参数引导进行假设检验,以及最近为机器学习应用程序生成数据。有许多序列模拟程序,但功能最丰富的程序往往运行速度较慢,而最快的程序往往功能较少。在这里,我们介绍 AliSim,这是一种新工具,可以在广泛的复杂进化模型下有效地模拟具有生物学意义的排列。为了在广泛的模拟条件下实现高性能,AliSim 实现了一种自适应方法,该方法结合了常用的速率矩阵和概率矩阵方法。AliSim 模拟一百万条序列或位点的对齐需要 1.4 小时和 1.3GB RAM,而流行的软件 Seq-Gen、Dawg 和 INDELible 需要 2-5 小时和 50-500GB 的 RAM。我们将 AliSim 作为 IQ-TREE 软件版本 2.2 的扩展提供,可在 www.iqtree.org 免费获得,并在 http://www.iqtree.org/doc/AliSim 上提供全面的用户教程。