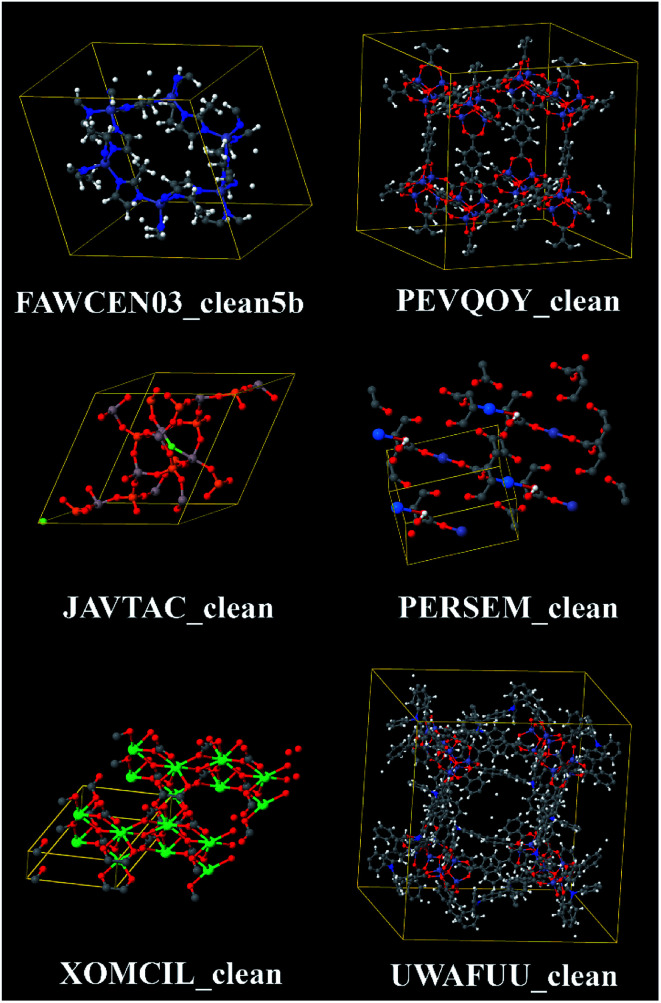
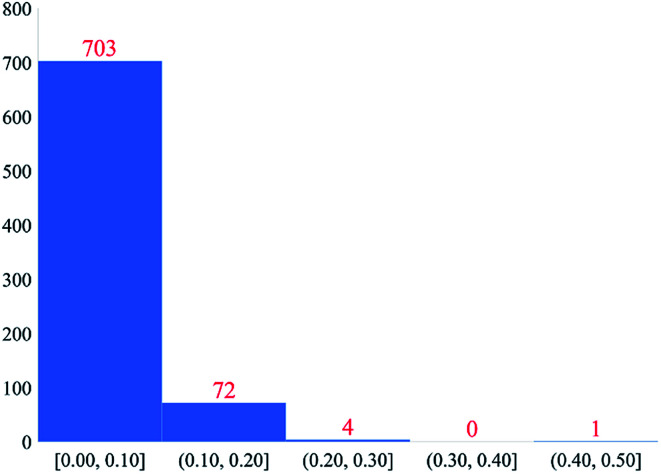
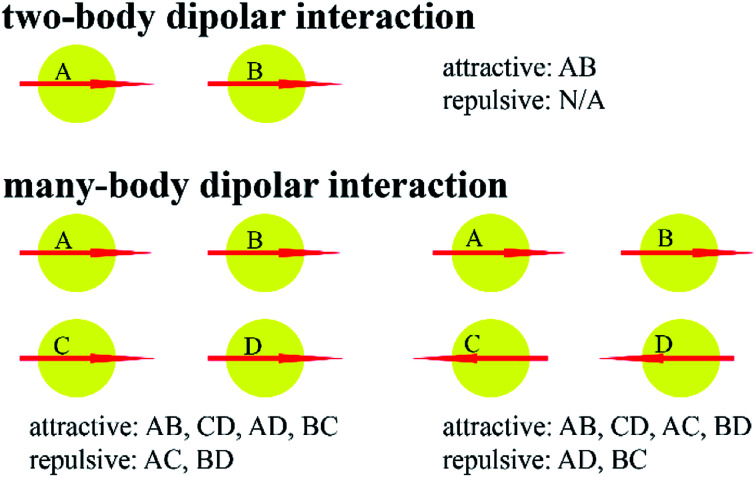
Chen Taoyi, Manz Thomas A
Department of Chemical & Materials Engineering, New Mexico State University Las Cruces New Mexico 88003-8001 USA
RSC Adv. 2019 Nov 13;9(63):36492-36507. doi: 10.1039/c9ra07327b. eCollection 2019 Nov 11.
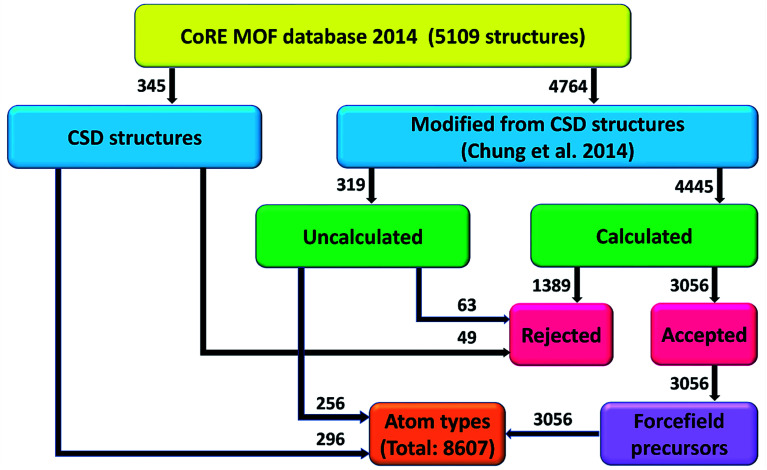
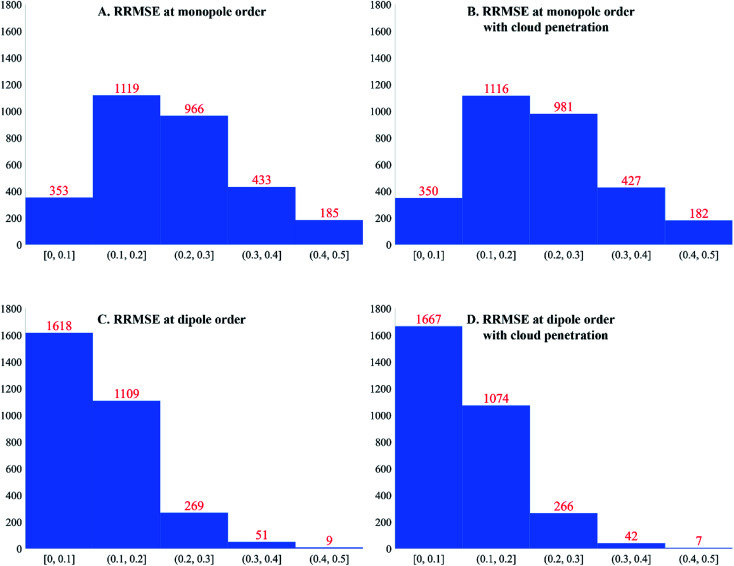
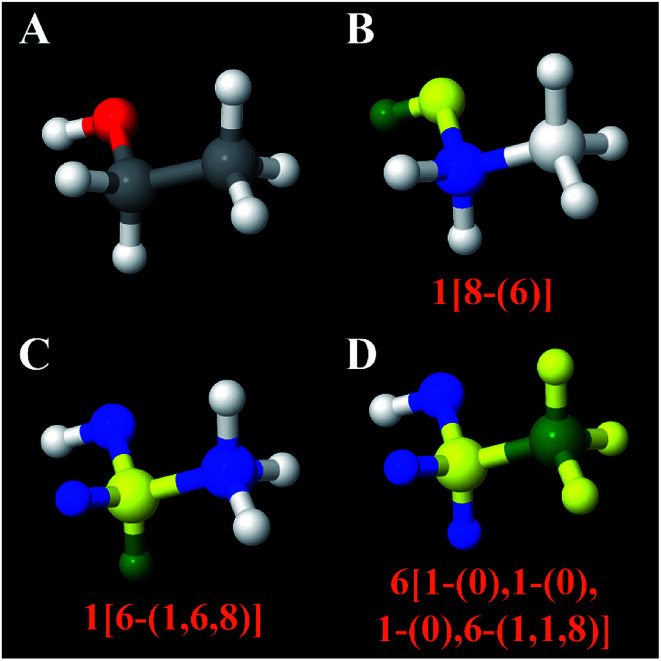
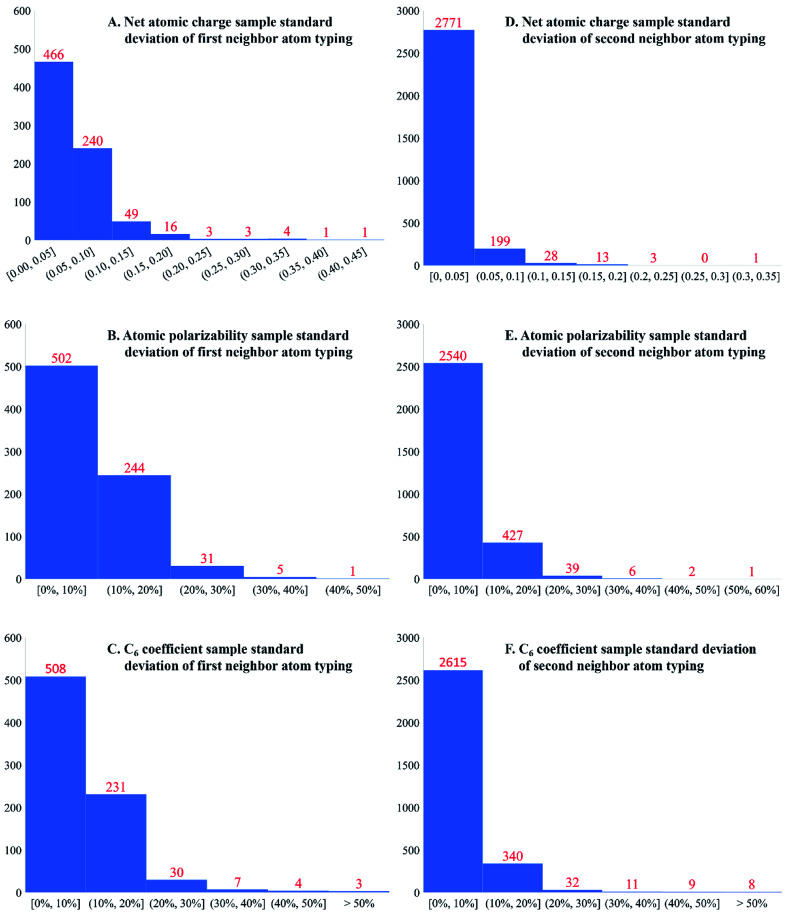
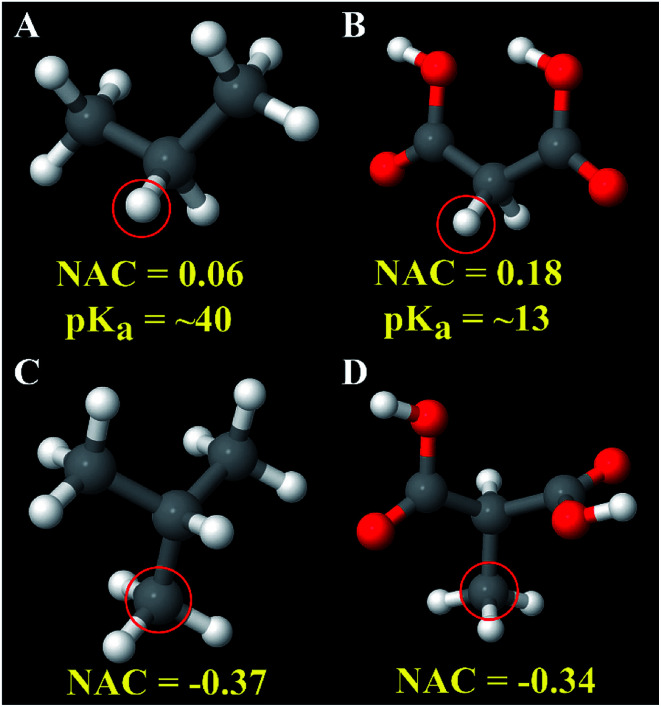
A host of important performance properties for metal-organic frameworks (MOFs) and other complex materials can be calculated by modeling statistical ensembles. The principle challenge is to develop accurate and computationally efficient interaction models for these simulations. Two major approaches are (i) molecular dynamics in which the interaction model is provided by an exchange-correlation theory (, DFT + dispersion functional) and (ii) molecular mechanics in which the interaction model is a parameterized classical force field. The first approach requires further development to improve computational speed. The second approach requires further development to automate accurate forcefield parameterization. Because of the extreme chemical diversity across thousands of MOF structures, this problem is still mostly unsolved today. For example, here we show structures in the 2014 CoRE MOF database contain more than 8 thousand different atom types based on first and second neighbors. Our results showed that atom types based on both first and second neighbors adequately capture the chemical environment, but atom types based on only first neighbors do not. For 3056 MOFs, we used density functional theory (DFT) followed by DDEC6 atomic population analysis to extract a host of important forcefield precursors: partial atomic charges; atom-in-material (AIM) C, C, and C dispersion coefficients; AIM dipole and quadrupole moments; various AIM polarizabilities; quantum Drude oscillator parameters; AIM electron cloud parameters; Electrostatic parameters were validated through comparisons to the DFT-computed electrostatic potential. These forcefield precursors should find widespread applications to developing MOF force fields.
通过对统计系综进行建模,可以计算金属有机框架(MOF)和其他复杂材料的一系列重要性能。主要挑战在于为这些模拟开发准确且计算效率高的相互作用模型。两种主要方法是:(i)分子动力学,其中相互作用模型由交换关联理论(如DFT + 色散泛函)提供;(ii)分子力学,其中相互作用模型是参数化的经典力场。第一种方法需要进一步发展以提高计算速度。第二种方法需要进一步发展以实现准确力场参数化的自动化。由于数千种MOF结构具有极大的化学多样性,这个问题至今大多仍未解决。例如,我们在此展示2014年CoRE MOF数据库中的结构基于第一和第二近邻包含八千多种不同的原子类型。我们的结果表明,基于第一和第二近邻的原子类型能够充分捕捉化学环境,但仅基于第一近邻的原子类型则不能。对于3056种MOF,我们使用密度泛函理论(DFT),随后进行DDEC6原子布居分析,以提取一系列重要的力场前体:部分原子电荷;材料中原子(AIM)的C、C和C色散系数;AIM偶极矩和四极矩;各种AIM极化率;量子德鲁德振子参数;AIM电子云参数;通过与DFT计算的静电势进行比较来验证静电参数。这些力场前体在开发MOF力场方面应会有广泛应用。