Du Pan, Zhao Jiyang
School of Life Science and Chemistry, Jiangsu Second Normal University Nanjing 210013 China.
School of Environmental Science, Nanjing Xiaozhuang University Nanjing 211171 China
RSC Adv. 2019 Nov 19;9(64):37675-37685. doi: 10.1039/c9ra07985h. eCollection 2019 Nov 13.
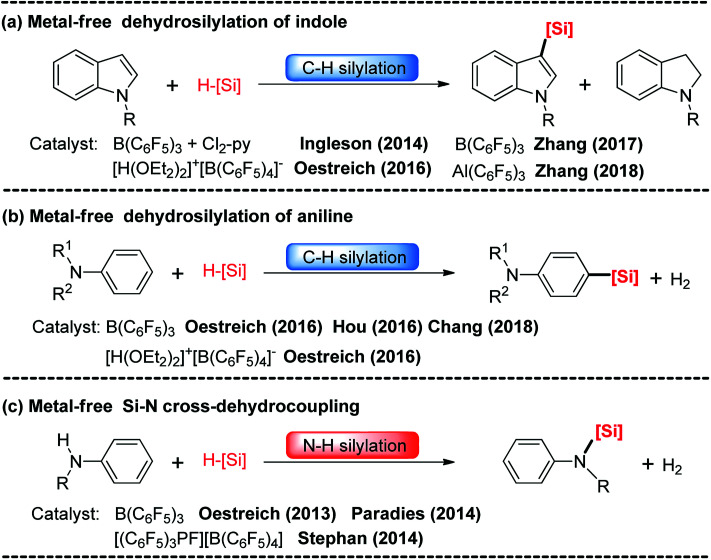
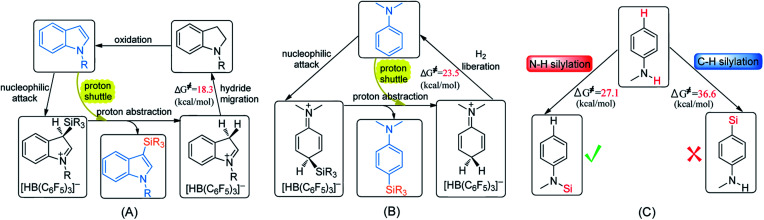
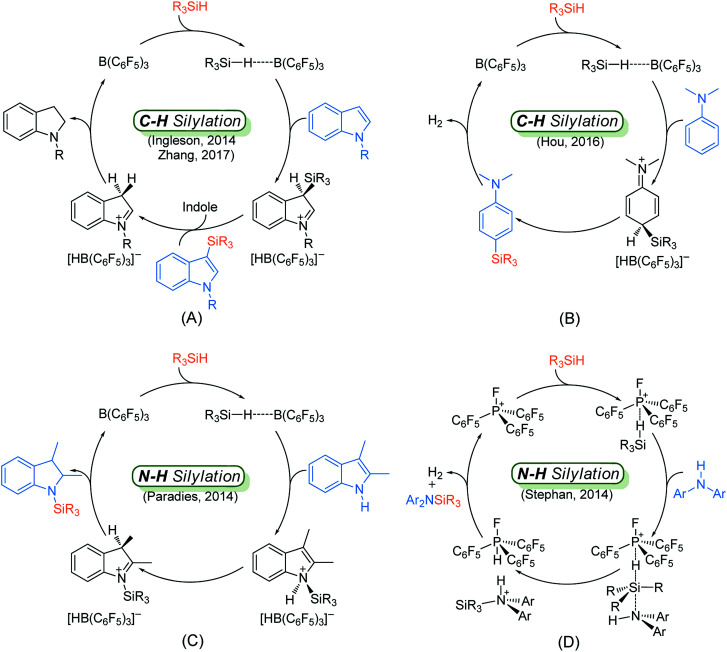
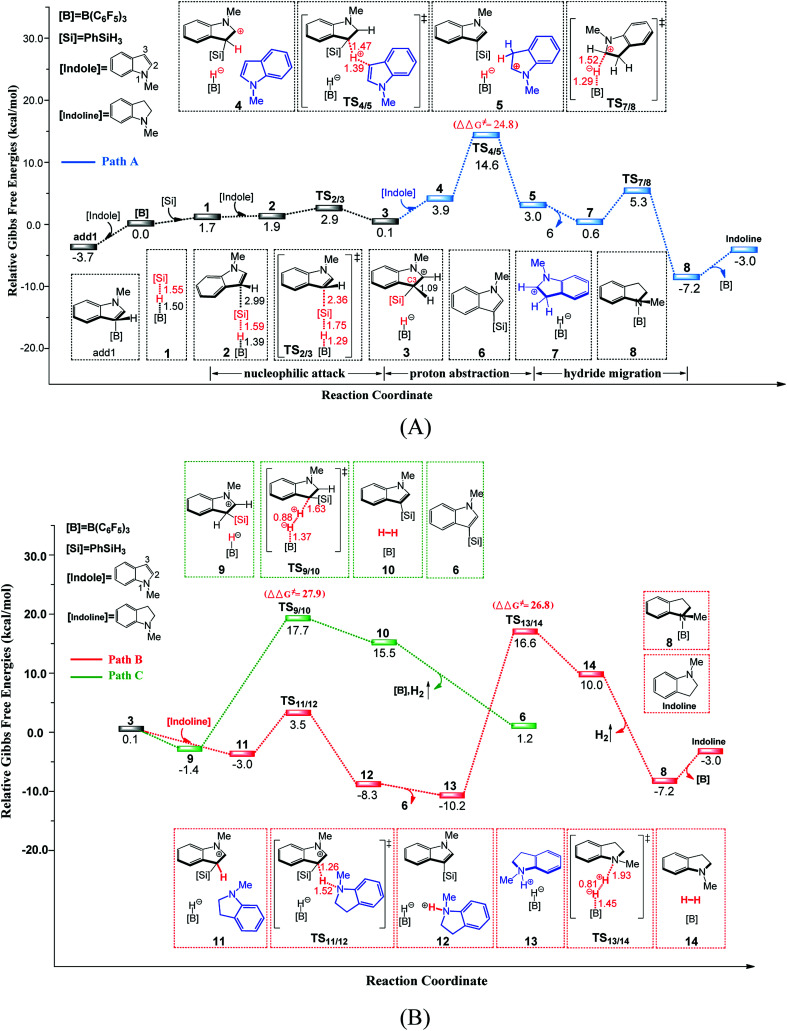
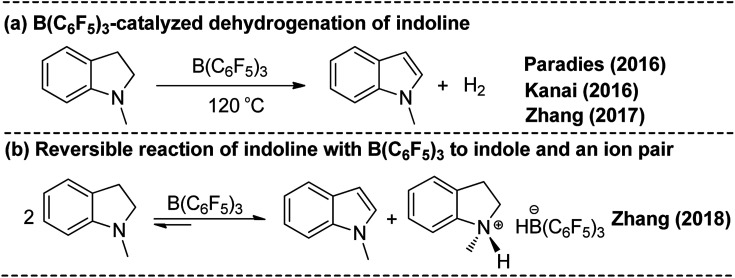
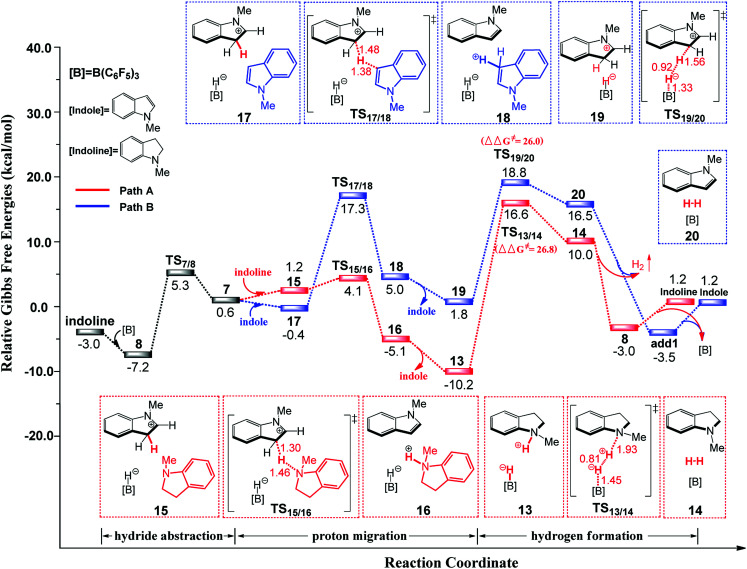
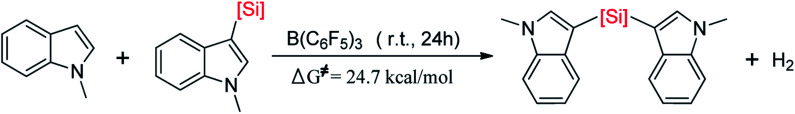
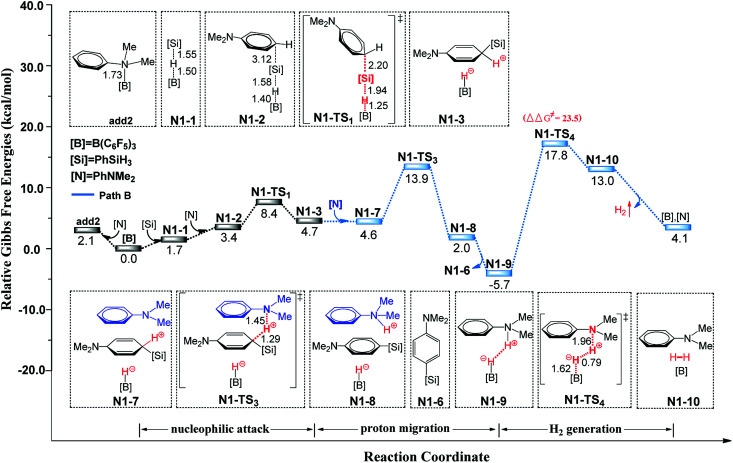
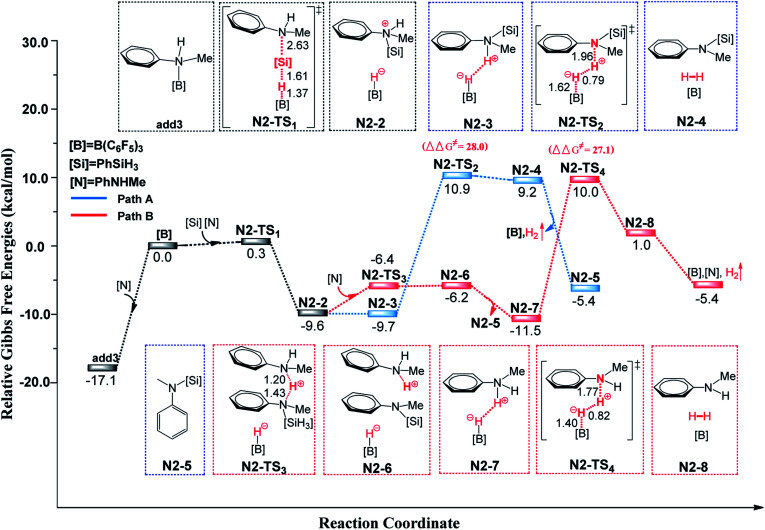
Direct selective dehydrogenative silylation of thiophenes, pyridines, indoles and anilines to synthesize silyl-substituted aromatic compounds catalyzed by metal-free Lewis acids was achieved recently. However, there is still insufficient mechanistic data for these transformations. Using density functional theory calculations, we conducted a detailed investigation of the mechanism of the B(CF)-catalyzed dehydrogenative silylation of -methylindole, ,-dimethylaniline and -methylaniline. We successfully located the most favourable reaction pathways that can explain the experimental observations notably well. The most favourable pathway for B(CF)-catalyzed C-H silylation of -methylindole includes nucleophilic attack, proton abstraction and hydride migration. The C-H silylation of ,-dimethylaniline follows a similar pathway to -methylindole rather than that proposed by Hou's group. Our mechanism successfully explains that the transformations of -methylindoline to -methylindole produce different products at different temperatures. For -methylaniline bearing both N-H and -phenyl C-H bonds, the N-H silylation reaction is more facile than the C-H silylation reaction. Our proposed mechanism of N-H silylation of -methylaniline is different from that proposed by the groups of Paradies and Stephan. Lewis acids Al(CF), Ga(CF) and B(2,6-ClCH)(-HCF) can also catalyze the C-H silylation of -methylindole like B(CF), but the most favourable pathways are those promoted by -methylindoline. Furthermore, we also found several other types of substrates that would undergo C-H or N-H silylation reactions under moderate conditions. These findings may facilitate the design of new catalysts for the dehydrogenative silylation of inactivated (hetero)arenes.
最近实现了在无金属路易斯酸催化下,噻吩、吡啶、吲哚和苯胺的直接选择性脱氢硅基化反应,以合成硅基取代的芳香族化合物。然而,这些转化反应的机理数据仍然不足。我们使用密度泛函理论计算,对B(CF)催化的α-甲基吲哚、N,N-二甲基苯胺和对甲基苯胺的脱氢硅基化反应机理进行了详细研究。我们成功找到了最有利的反应途径,能很好地解释实验结果。B(CF)催化α-甲基吲哚的C-H硅基化反应的最有利途径包括亲核进攻、质子夺取和氢化物迁移。N,N-二甲基苯胺的C-H硅基化反应遵循与α-甲基吲哚相似的途径,而不是Hou课题组提出的途径。我们的机理成功解释了α-甲基二氢吲哚向α-甲基吲哚的转化在不同温度下会产生不同产物。对于同时含有N-H和对位苯基C-H键的对甲基苯胺,N-H硅基化反应比C-H硅基化反应更容易进行。我们提出的对甲基苯胺N-H硅基化反应的机理与Paradies和Stephan课题组提出的不同。路易斯酸Al(CF)、Ga(CF)和B(2,6-Cl₂C₆H₃)(-HCF₂)也能像B(CF)一样催化α-甲基吲哚的C-H硅基化反应,但最有利的途径是由α-甲基二氢吲哚促进的途径。此外,我们还发现了其他几种类型的底物,它们在温和条件下会发生C-H或N-H硅基化反应。这些发现可能有助于设计用于失活(杂)芳烃脱氢硅基化反应的新型催化剂。