Murakawa Takeshi, Kurihara Kazuo, Adachi Motoyasu, Kusaka Katsuhiro, Tanizawa Katsuyuki, Okajima Toshihide
Department of Biochemistry, Faculty of Medicine, Osaka Medical and Pharmaceutical University, 2-7 Daigakumachi, Takatsuki, Osaka 569-8686, Japan.
Institute for Quantum Life Science, Quantum Life and Medical Science Directorate, National Institutes for Quantum Science and Technology, 2-4 Shirakata, Tokai, Ibaraki 319-1106, Japan.
IUCrJ. 2022 Apr 8;9(Pt 3):342-348. doi: 10.1107/S2052252522003657. eCollection 2022 May 1.
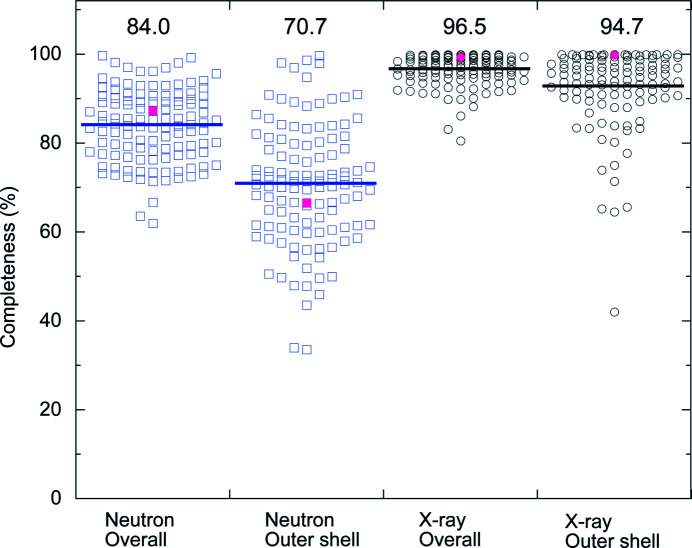
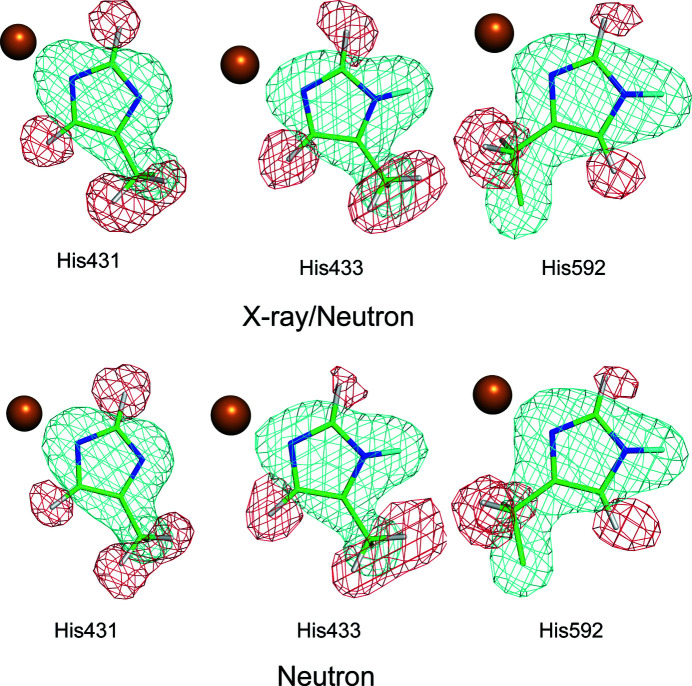
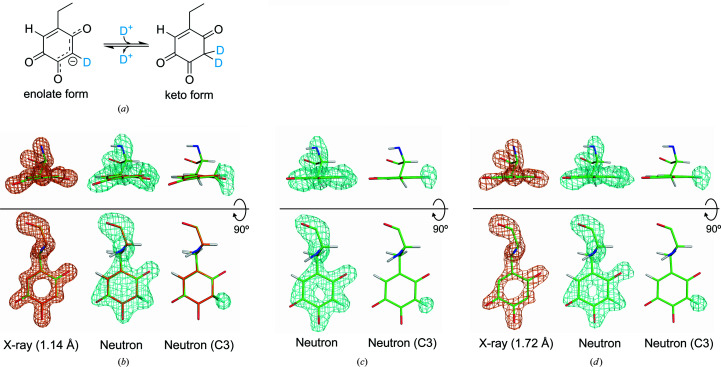
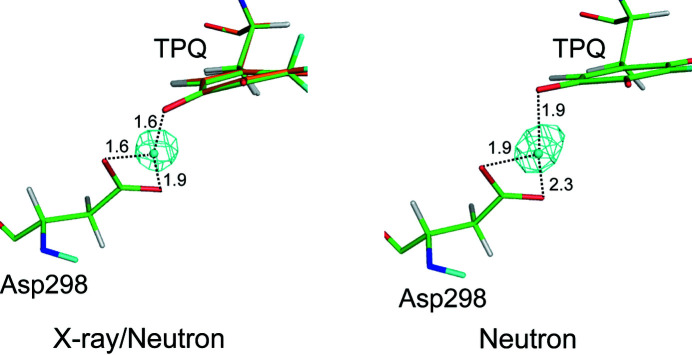
Protein neutron crystallography is a powerful technique to determine the positions of H atoms, providing crucial biochemical information such as the protonation states of catalytic groups and the geometry of hydrogen bonds. Recently, the crystal structure of a bacterial copper amine oxidase was determined by joint refinement using X-ray and neutron diffraction data sets at resolutions of 1.14 and 1.72 Å, respectively [Murakawa (2020 ▸). , , 10818-10824]. While joint refinement is effective for the determination of the accurate positions of heavy atoms on the basis of the electron density, the structural information on light atoms (hydrogen and deuterium) derived from the neutron diffraction data might be affected by the X-ray data. To unravel the information included in the neutron diffraction data, the structure determination was conducted again using only the neutron diffraction data at 1.72 Å resolution and the results were compared with those obtained in the previous study. Most H and D atoms were identified at essentially the same positions in both the neutron-only and the X-ray/neutron joint refinements. Nevertheless, neutron-only refinement was found to be less effective than joint refinement in providing very accurate heavy-atom coordinates that lead to significant improvement of the neutron scattering length density map, especially for the active-site cofactor. Consequently, it was confirmed that X-ray/neutron joint refinement is crucial for determination of the real chemical structure of the catalytic site of the enzyme.
蛋白质中子晶体学是确定氢原子位置的一种强大技术,能提供关键的生化信息,如催化基团的质子化状态和氢键的几何结构。最近,通过分别使用分辨率为1.14 Å和1.72 Å的X射线和中子衍射数据集进行联合精修,确定了一种细菌铜胺氧化酶的晶体结构[村川(2020▸)。,,10818 - 10824]。虽然联合精修对于基于电子密度确定重原子的准确位置是有效的,但从中子衍射数据得出的轻原子(氢和氘)的结构信息可能会受到X射线数据的影响。为了解开中子衍射数据中包含的信息,仅使用1.72 Å分辨率的中子衍射数据再次进行结构测定,并将结果与先前研究中获得的结果进行比较。在仅中子精修和X射线/中子联合精修中,大多数氢和氘原子在基本相同的位置被识别出来。然而,发现仅中子精修在提供非常精确的重原子坐标方面不如联合精修有效,而这些重原子坐标会显著改善中子散射长度密度图,特别是对于活性位点辅因子。因此,证实了X射线/中子联合精修对于确定该酶催化位点的真实化学结构至关重要。