Invitae Corporation, San Francisco, California, USA.
Indiana University Health, Indianapolis, Indiana, USA.
Am J Med Genet A. 2022 Sep;188(9):2642-2651. doi: 10.1002/ajmg.a.62779. Epub 2022 May 16.
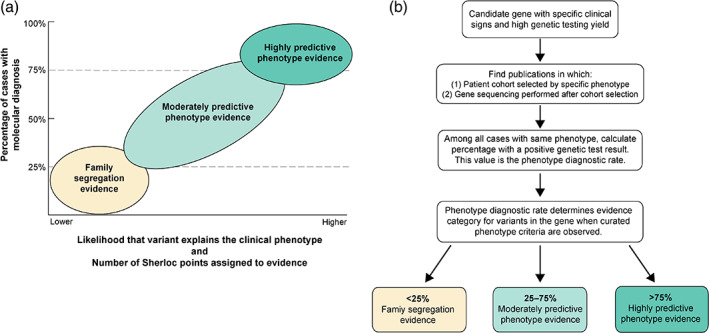
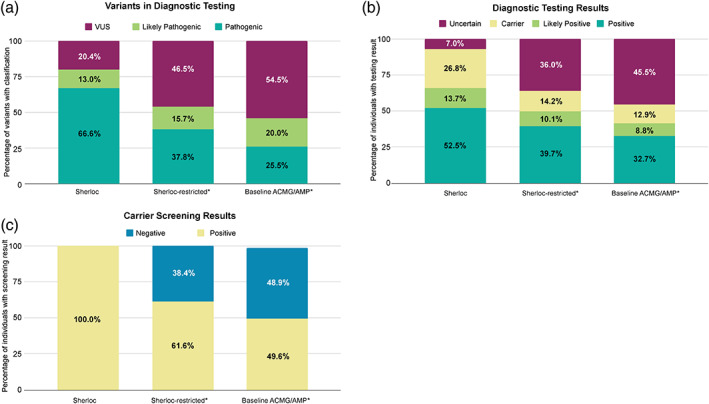
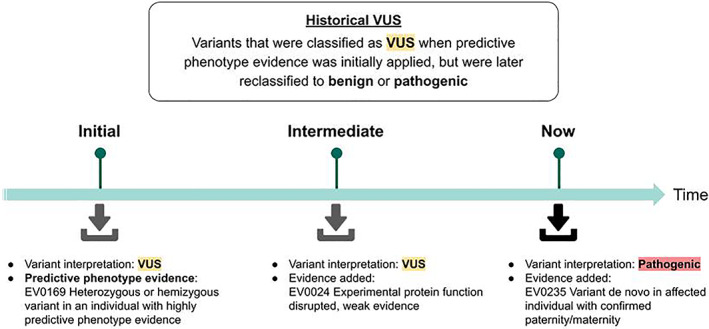
Guidelines for variant interpretation include criteria for incorporating phenotype evidence, but this evidence is inconsistently applied. Systematic approaches to using phenotype evidence are needed. We developed a method for curating disease phenotypes as highly or moderately predictive of variant pathogenicity based on the frequency of their association with disease-causing variants. To evaluate this method's accuracy, we retrospectively reviewed variants with clinical classifications that had evolved from uncertain to definitive in genes associated with curated predictive phenotypes. To demonstrate the clinical validity and utility of this approach, we compared variant classifications determined with and without predictive phenotype evidence. The curation method was accurate for 93%-98% of eligible variants. Among variants interpreted using highly predictive phenotype evidence, the percentage classified as pathogenic or likely pathogenic was 80%, compared with 46%-54% had the evidence not been used. Positive results among individuals harboring variants with highly predictive phenotype-guided interpretations would have been missed in 25%-37% of diagnostic tests and 39%-50% of carrier screens had other approaches to phenotype evidence been used. In summary, predictive phenotype evidence associated with specific curated genes can be systematically incorporated into variant interpretation to reduce uncertainty and increase the clinical utility of genetic testing.
变异解释指南包括纳入表型证据的标准,但这些证据的应用并不一致。需要系统的方法来利用表型证据。我们开发了一种方法,根据与致病变异相关的疾病表型的关联频率,对高度或中度预测变异致病性的疾病表型进行编目。为了评估该方法的准确性,我们回顾性地审查了与编目预测表型相关的基因中,其临床分类从不确定转变为明确的变异。为了证明该方法的临床有效性和实用性,我们比较了有和没有预测表型证据的变异分类。编目方法对 93%-98%的合格变异是准确的。在使用高度预测表型证据进行解释的变异中,被归类为致病性或可能致病性的比例为 80%,而如果不使用该证据,则为 46%-54%。如果使用了其他表型证据的方法,在携带高度预测表型指导解释的变异的个体中,阳性结果将在 25%-37%的诊断测试和 39%-50%的携带者筛查中被遗漏。总之,与特定编目基因相关的预测表型证据可以系统地纳入变异解释,以减少不确定性并提高遗传检测的临床实用性。