AquaMem Consultants, Rodeo, New Mexico, NM 88056, USA.
Institute for Sustainable Industries and Liveable Cities, Victoria University, Melbourne, VIC 3030, Australia.
Viruses. 2022 May 11;14(5):1029. doi: 10.3390/v14051029.
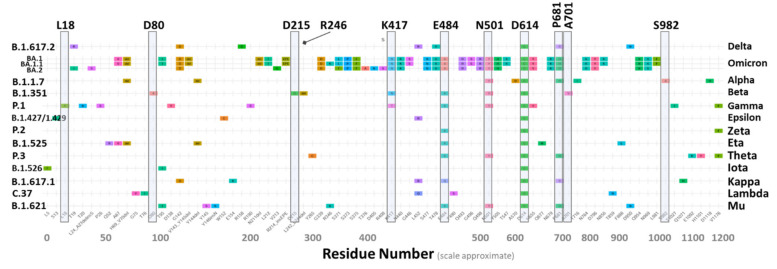
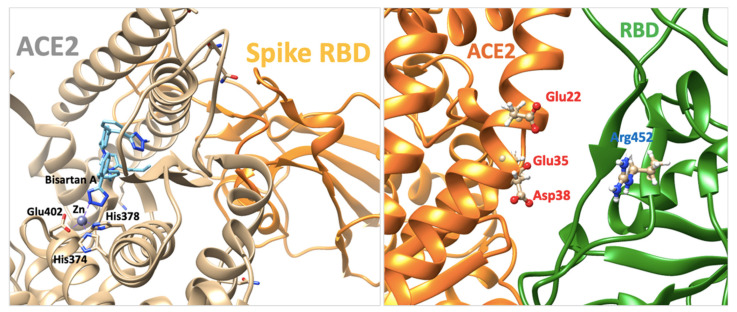
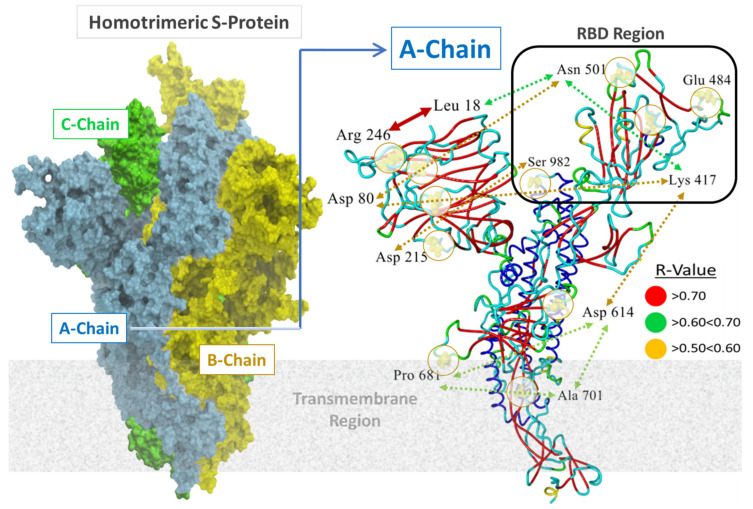
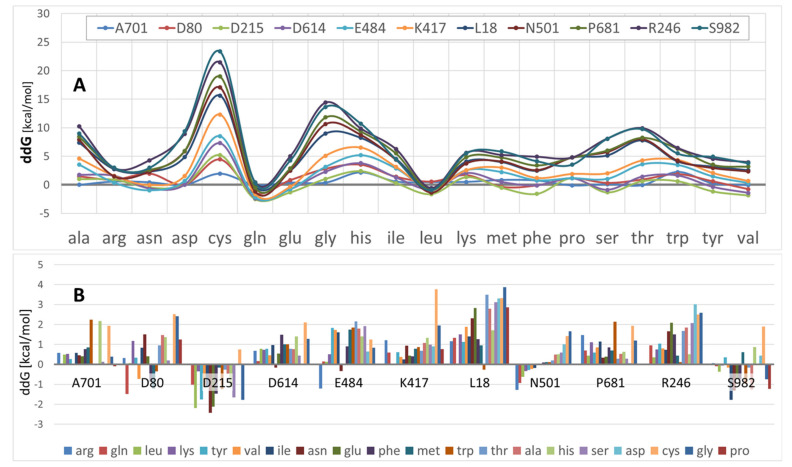
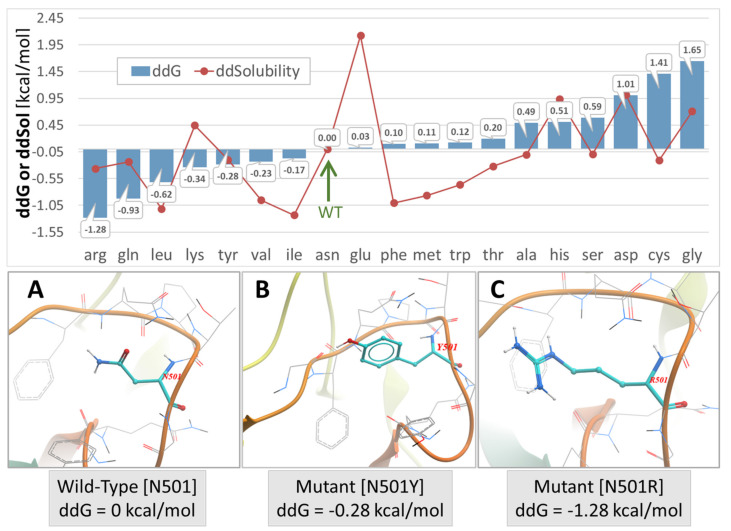
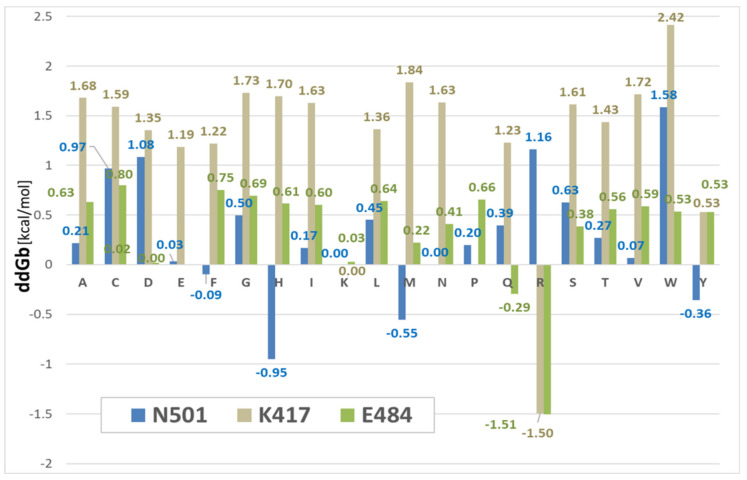
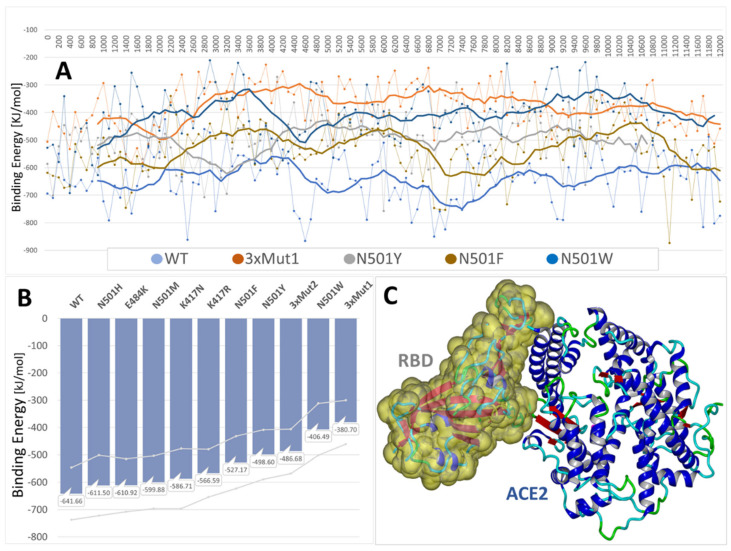
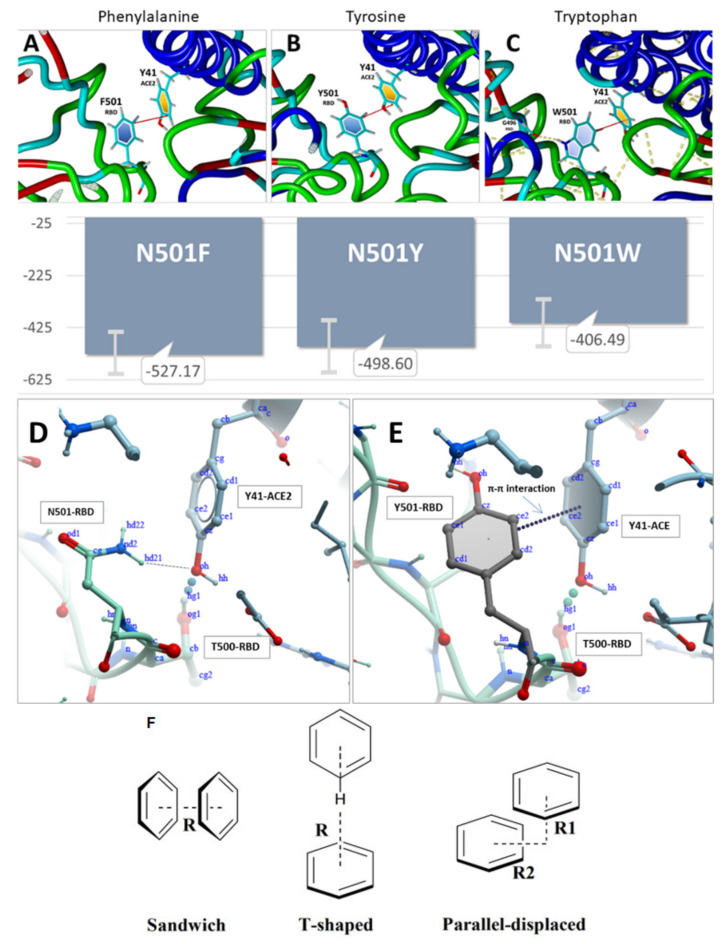
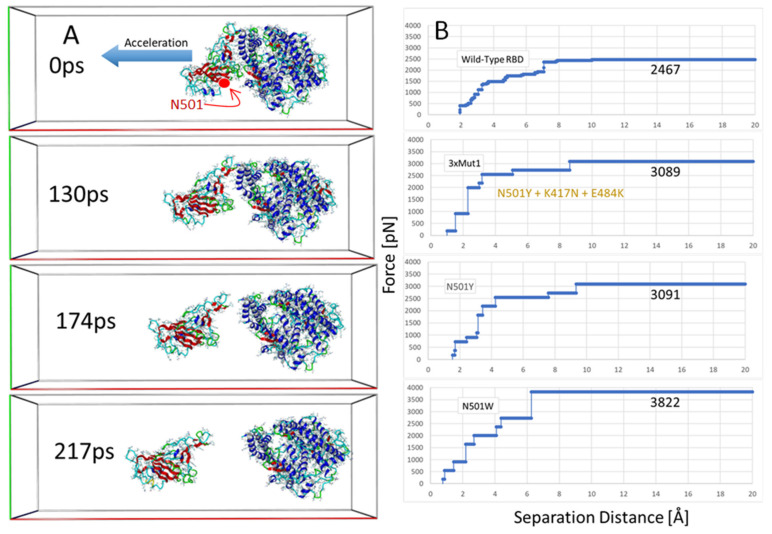
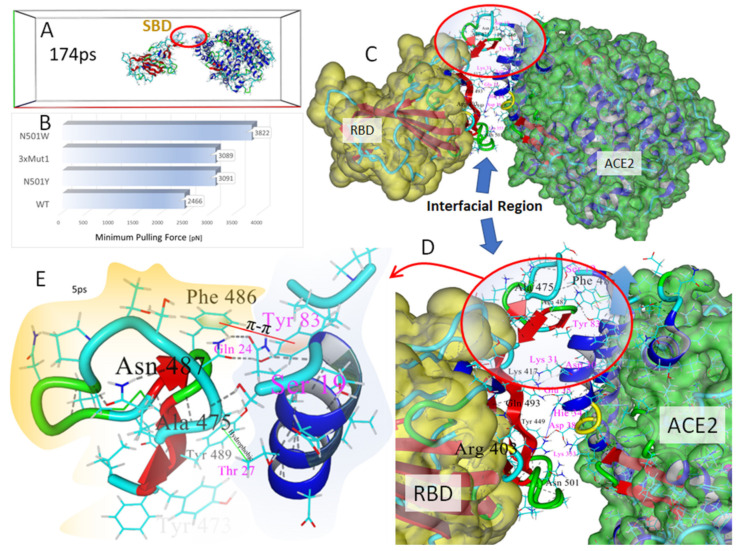
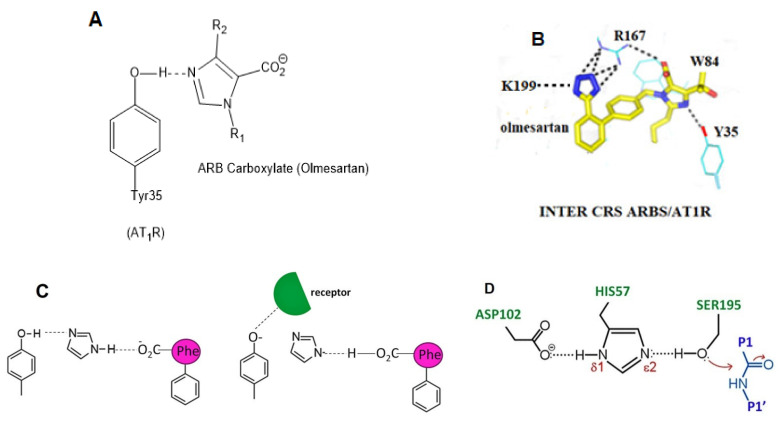
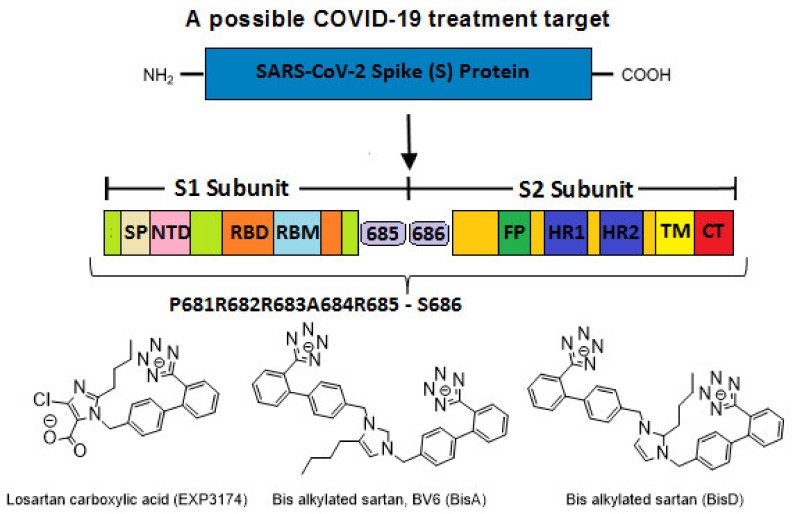
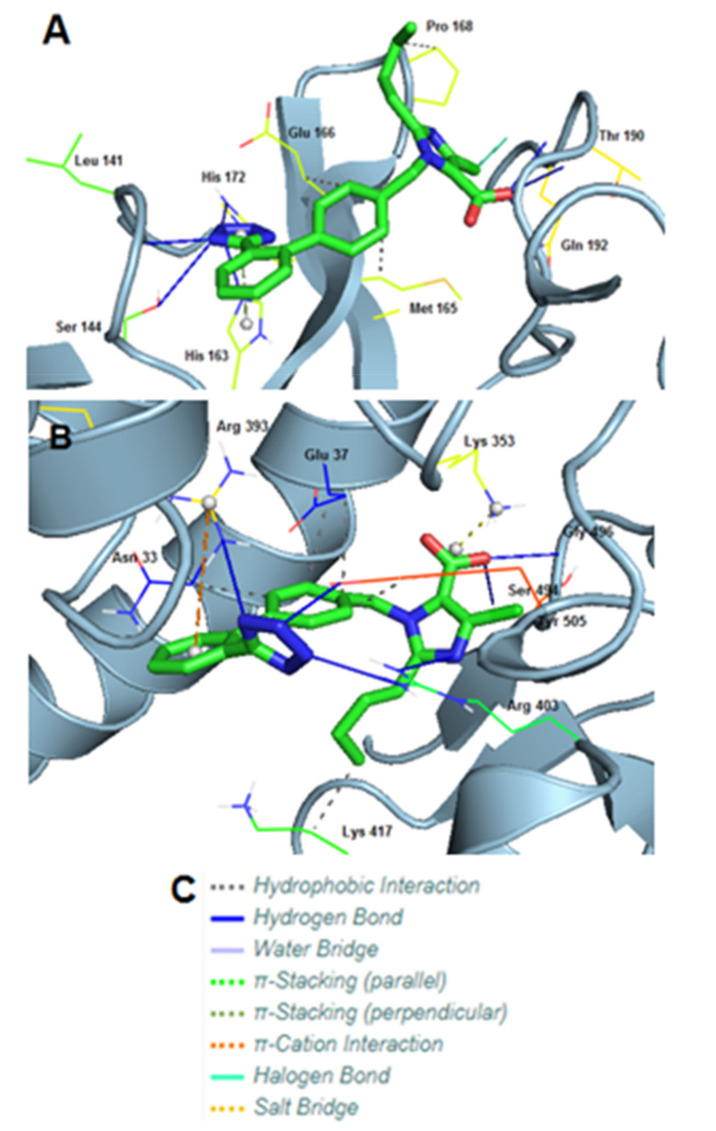
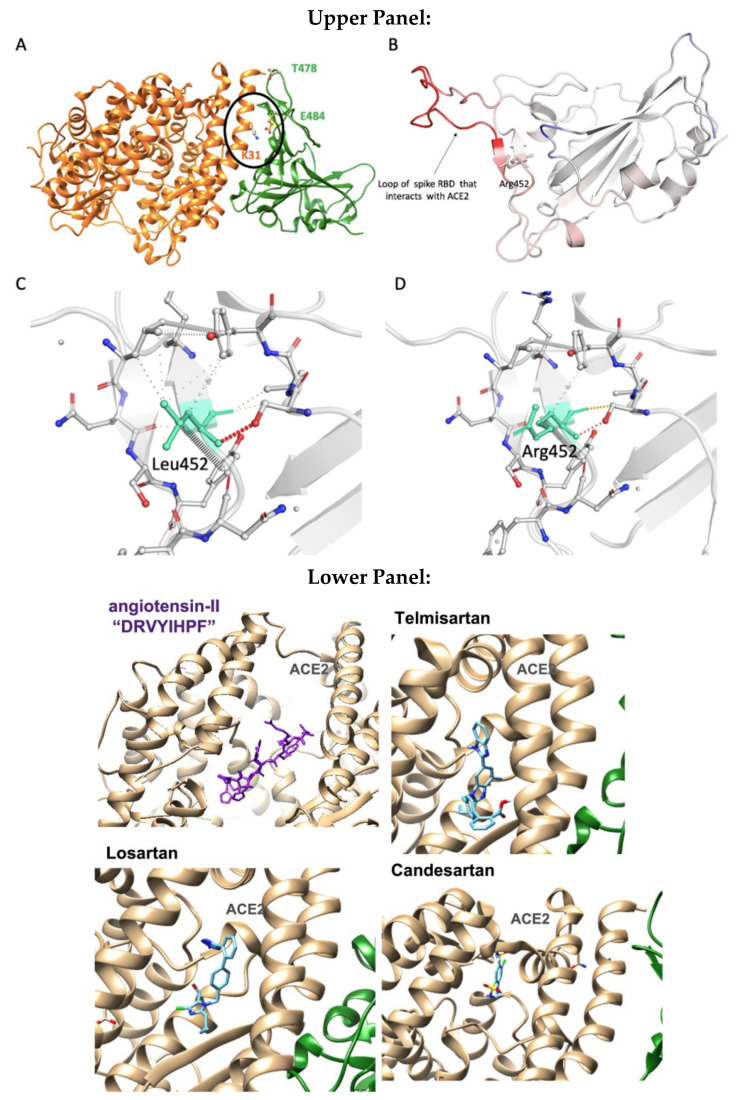
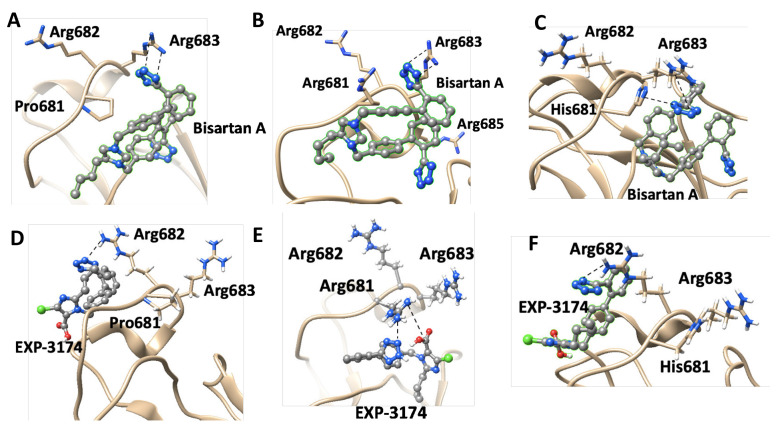
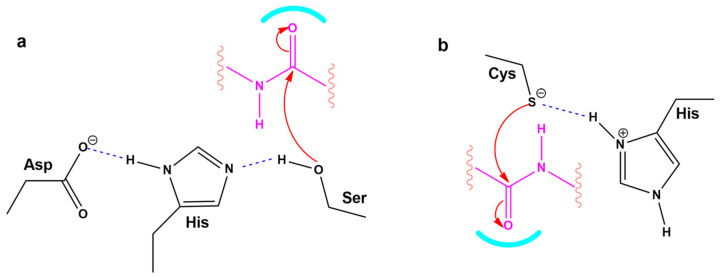
SARS-CoV-2 is a global challenge due to its ability to mutate into variants that spread more rapidly than the wild-type virus. Because the molecular biology of this virus has been studied in such great detail, it represents an archetypal paradigm for research into new antiviral drug therapies. The rapid evolution of SARS-CoV-2 in the human population is driven, in part, by mutations in the receptor-binding domain (RBD) of the spike (S-) protein, some of which enable tighter binding to angiotensin-converting enzyme (ACE2). More stable RBD-ACE2 association is coupled with accelerated hydrolysis of furin and 3CLpro cleavage sites that augment infection. Non-RBD and non-interfacial mutations assist the S-protein in adopting thermodynamically favorable conformations for stronger binding. The driving forces of key mutations for Alpha, Beta, Gamma, Delta, Kappa, Lambda and Omicron variants, which stabilize the RBD-ACE2 complex, are investigated by free-energy computational approaches, as well as equilibrium and steered molecular dynamic simulations. Considered also are the structural hydropathy traits of the residues in the interface between SARS-CoV-2 RBD and ACE2 protein. Salt bridges and π-π interactions are critical forces that create stronger complexes between the RBD and ACE2. The trend of mutations is the replacement of non-polar hydrophobic interactions with polar hydrophilic interactions, which enhance binding of RBD with ACE2. However, this is not always the case, as conformational landscapes also contribute to a stronger binding. Arginine, the most polar and hydrophilic among the natural amino acids, is the most aggressive mutant amino acid for stronger binding. Arginine blockers, such as traditional sartans that bear anionic tetrazoles and carboxylates, may be ideal candidate drugs for retarding viral infection by weakening S-protein RBD binding to ACE2 and discouraging hydrolysis of cleavage sites. Based on our computational results it is suggested that a new generation of "supersartans", called "bisartans", bearing two anionic biphenyl-tetrazole pharmacophores, are superior to carboxylates in terms of their interactions with viral targets, suggesting their potential as drugs in the treatment of COVID-19. In Brief: This in silico study reviews our understanding of molecular driving forces that trigger mutations in the SARS-CoV-2 virus. It also reports further studies on a new class of "supersartans" referred to herein as "bisartans", bearing two anionic biphenyltetrazole moieties that show potential in models for blocking critical amino acids of mutants, such as arginine, in the Delta variant. Bisartans may also act at other targets essential for viral infection and replication (i.e., ACE2, furin cleavage site and 3CLpro), rendering them potential new drugs for additional experimentation and translation to human clinical trials.
SARS-CoV-2 是一种全球性的挑战,因为它能够突变为传播速度比野生型病毒更快的变体。由于对这种病毒的分子生物学进行了如此深入的研究,它代表了研究新的抗病毒药物治疗方法的典型范例。SARS-CoV-2 在人群中的快速进化部分是由于刺突(S-)蛋白的受体结合域(RBD)中的突变引起的,其中一些突变使病毒能够更紧密地与血管紧张素转换酶(ACE2)结合。更稳定的 RBD-ACE2 结合伴随着弗林和 3CLpro 切割位点水解的加速,从而增强了感染。非 RBD 和非界面突变有助于 S 蛋白采用更有利于结合的热力学有利构象。通过自由能计算方法、平衡和导向分子动力学模拟研究了 Alpha、Beta、Gamma、Delta、Kappa、Lambda 和 Omicron 变体中关键突变的驱动因素,这些变体稳定了 RBD-ACE2 复合物。还考虑了 SARS-CoV-2 RBD 和 ACE2 蛋白之间界面残基的结构疏水性特征。盐桥和π-π 相互作用是在 RBD 和 ACE2 之间形成更强复合物的关键力。突变的趋势是用极性亲水相互作用取代非极性疏水相互作用,从而增强 RBD 与 ACE2 的结合。然而,情况并非总是如此,因为构象景观也有助于更强的结合。精氨酸是天然氨基酸中最极性和最亲水的,是用于更强结合的最具侵略性的突变氨基酸。精氨酸阻滞剂,如带有阴离子四唑和羧酸的传统沙坦类药物,可能是通过削弱 S 蛋白 RBD 与 ACE2 的结合并阻止切割位点的水解来减缓病毒感染的理想候选药物。基于我们的计算结果,建议开发新一代称为“双沙坦”的“超级沙坦”,它带有两个阴离子联苯四唑药效团,在与病毒靶标相互作用方面优于羧酸,这表明它们有潜力成为治疗 COVID-19 的药物。简而言之:这项计算机研究综述了我们对引发 SARS-CoV-2 病毒突变的分子驱动力的理解。它还报告了对一类新的“超级沙坦”的进一步研究,称为“双沙坦”,它带有两个阴离子联苯四唑部分,在阻断 Delta 变体中关键氨基酸(如精氨酸)的模型中显示出潜力。双沙坦还可能作用于其他对病毒感染和复制至关重要的靶标(即 ACE2、弗林切割位点和 3CLpro),使其成为进一步实验和转化为人类临床试验的潜在新药。