Department of Biostatistics and Informatics, University of Colorado, Anschutz Medical Campus, Aurora, CO, USA.
Center for Genes, Environment and Health, National Jewish Health, 1400 Jackson St, Denver, 80206, CO, USA.
BMC Med Res Methodol. 2022 May 28;22(1):153. doi: 10.1186/s12874-022-01615-8.
As the cost of RNA-sequencing decreases, complex study designs, including paired, longitudinal, and other correlated designs, become increasingly feasible. These studies often include multiple hypotheses and thus multiple degree of freedom tests, or tests that evaluate multiple hypotheses jointly, are often useful for filtering the gene list to a set of interesting features for further exploration while controlling the false discovery rate. Though there are several methods which have been proposed for analyzing correlated RNA-sequencing data, there has been little research evaluating and comparing the performance of multiple degree of freedom tests across methods.
We evaluated 11 different methods for modelling correlated RNA-sequencing data by performing a simulation study to compare the false discovery rate, power, and model convergence rate across several hypothesis tests and sample size scenarios. We also applied each method to a real longitudinal RNA-sequencing dataset.
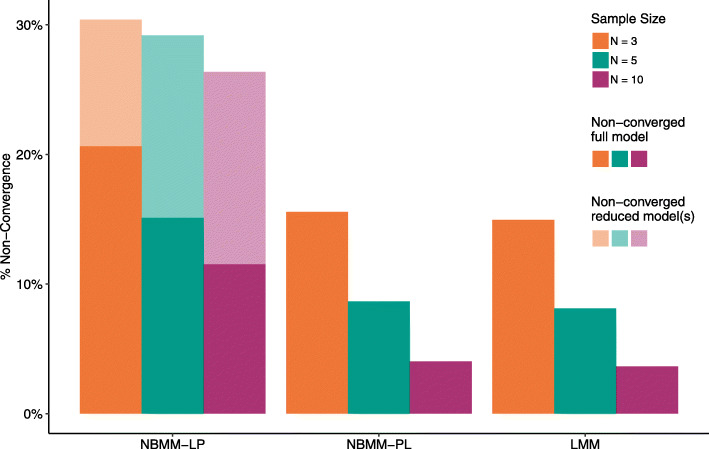
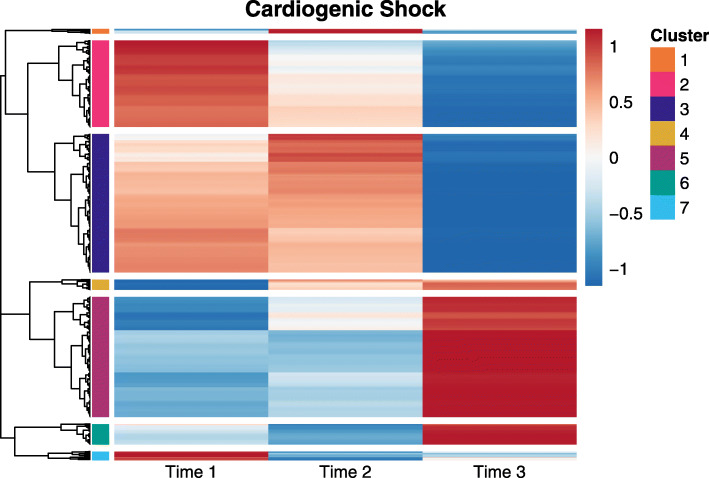
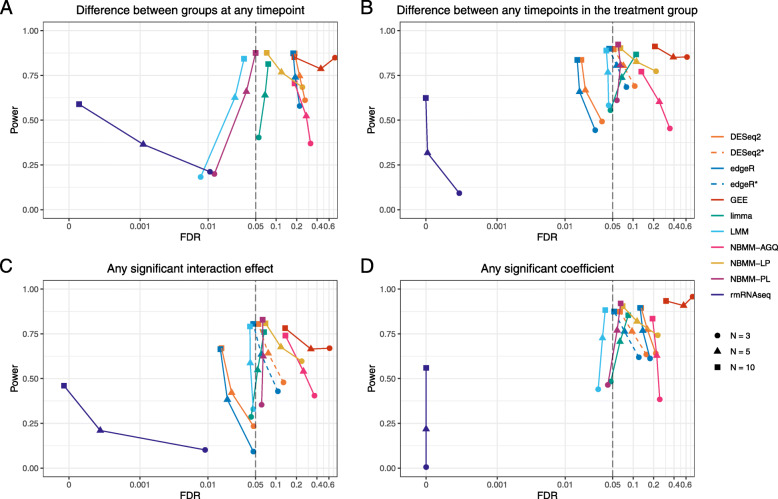
Linear mixed modelling using transformed data had the best false discovery rate control while maintaining relatively high power. However, this method had high model non-convergence, particularly at small sample sizes. No method had high power at the lowest sample size. We found a mix of conservative and anti-conservative behavior across the other methods, which was influenced by the sample size and the hypothesis being evaluated. The patterns observed in the simulation study were largely replicated in the analysis of a longitudinal study including data from intensive care unit patients experiencing cardiogenic or septic shock.
Multiple degree of freedom testing is a valuable tool in longitudinal and other correlated RNA-sequencing experiments. Of the methods that we investigated, linear mixed modelling had the best overall combination of power and false discovery rate control. Other methods may also be appropriate in some scenarios.
随着 RNA 测序成本的降低,包括配对、纵向和其他相关设计在内的复杂研究设计变得越来越可行。这些研究通常包含多个假设,因此多个自由度检验,或联合评估多个假设的检验,通常有助于将基因列表过滤到一组有趣的特征,以便进一步探索,同时控制假发现率。虽然已经提出了几种用于分析相关 RNA 测序数据的方法,但很少有研究评估和比较多种自由度检验在不同方法中的性能。
我们通过进行模拟研究来评估 11 种不同的方法,以比较几种假设检验和样本量场景下的假发现率、功效和模型收敛率。我们还将每种方法应用于真实的纵向 RNA 测序数据集。
使用转换后数据的线性混合建模具有最佳的假发现率控制,同时保持相对较高的功效。然而,该方法的模型非收敛性较高,特别是在样本量较小的情况下。在最小样本量下,没有一种方法具有高功效。我们发现,在其他方法中存在保守和反保守行为的混合,这受样本量和正在评估的假设的影响。在模拟研究中观察到的模式在对包括经历心源性或感染性休克的重症监护病房患者数据的纵向研究的分析中得到了很大程度的复制。
在纵向和其他相关 RNA 测序实验中,多个自由度检验是一种很有价值的工具。在所研究的方法中,线性混合建模具有最佳的功效和假发现率控制的整体组合。其他方法在某些情况下也可能适用。