Xing Baobao, Shi Lei, Bao Zhiguo, Liang Ying, Liu Bo, Liu Ruihan
Department of Clinical Laboratory, Inner Mongolia Autonomous Region People's Hospital, Hohhot, China.
Department of Clinical Laboratory, PLA Rocket Force Characteristic Medical Center, Beijing, China.
J Thorac Dis. 2022 May;14(5):1638-1650. doi: 10.21037/jtd-22-557.
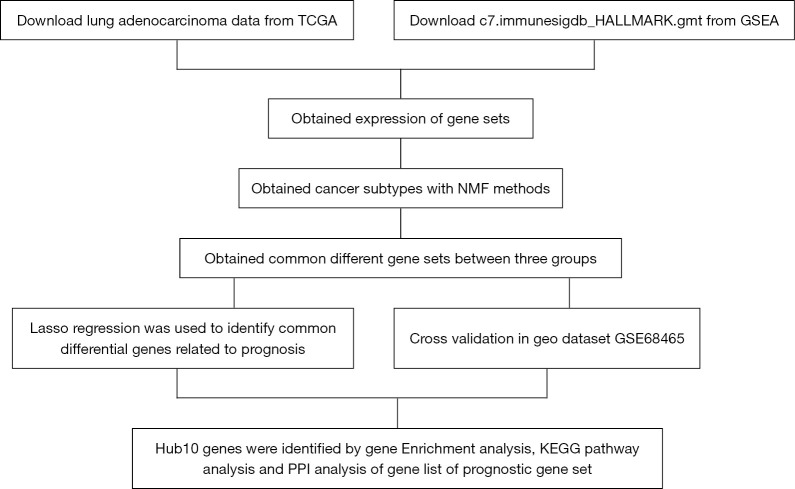
Lung adenocarcinoma (LUAD) is a subtype of lung cancer with high morbidity and mortality. While genotyping is an important determinant for the prognosis of LUAD patients, there is a paucity of studies on gene set-based expression (GSE) typing for LUAD. This current study used GSE methodology to perform gene typing of LUAD patients.
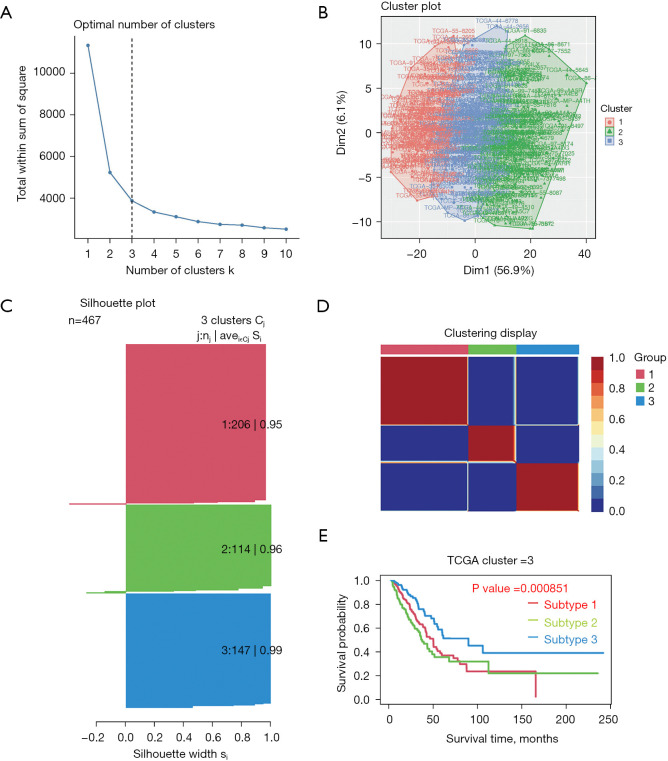
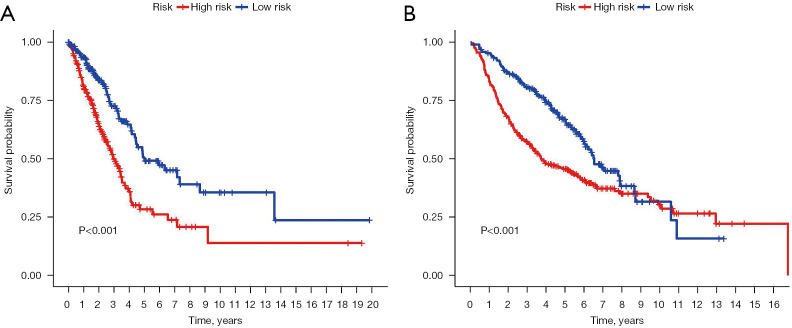
Clinical and genomic information of the LUAD patients were downloaded from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. Patients with LUAD were clustered into different molecular subtypes depending on the clinical and gene set expression characteristics. The survival rate and silhouette widths were compared between each molecular subtype. Differences in survival rate between gene sets were analyzed using Kaplan-Meier survival curves. Cox regression and Lasso regression were used to establish the prognostic gene set model based on the TCGA database, and the results were validated using the GEO dataset.
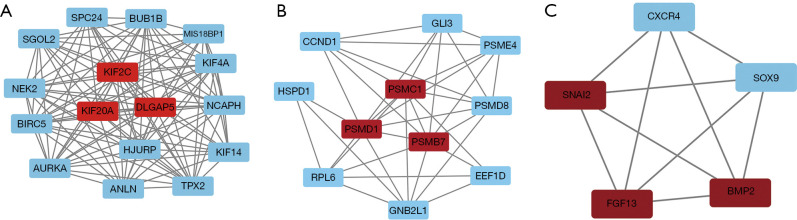
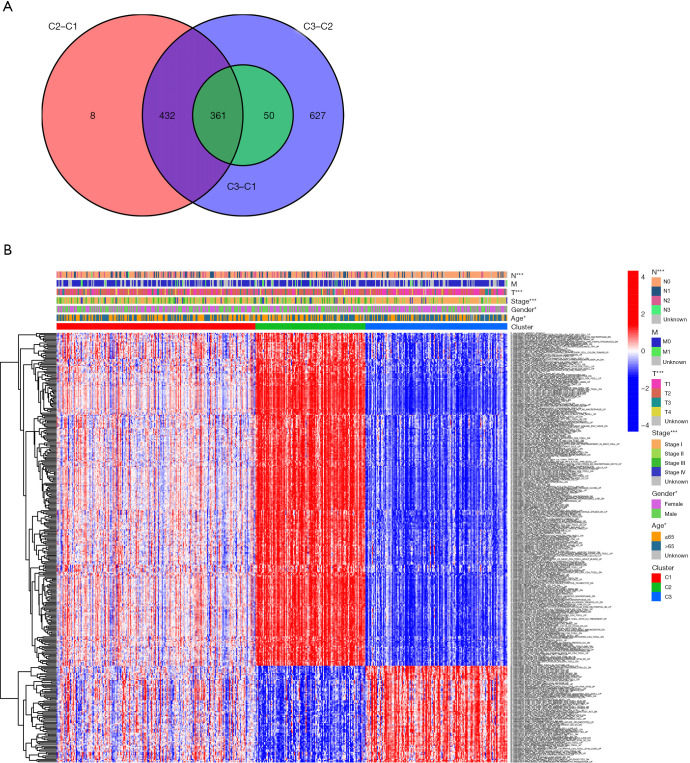
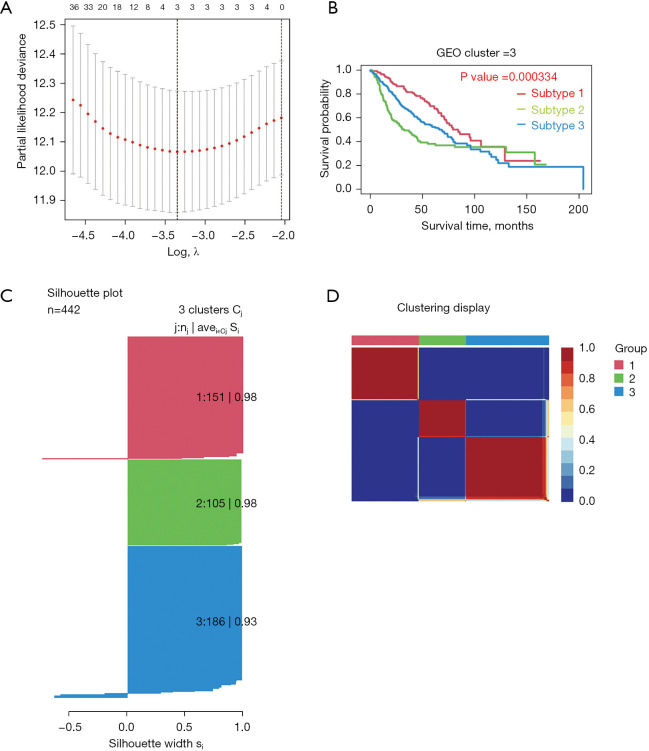
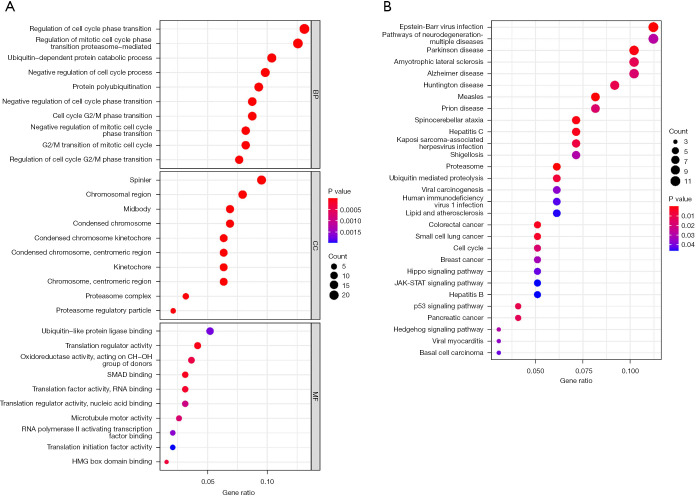
A total of 10 hub genes were finally identified and clustered into 3 subtypes with a mean contour width of 0.96. There were significant differences in survival rates among the 3 subtypes (P<0.05). Gene Ontology (GO) analysis indicated that the related biological processes (BP) were mainly involved in regulation of cell cycle, mitotic cell cycle phase transition, and proteasome-mediated ubiquitin-dependent protein catabolic process. The cellular components (CC) were related to the spindle, chromosomal region, and midbody. Molecular function (MF) mainly focused on ubiquitin-like protein ligase binding, translation regulator activity, and oxidation activity. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that the main pathways included the Epstein Barr virus infection pathway of neurogeneration, the p53 signaling pathway, and the proteome pathways. In addition, the protein-protein interaction network was analyzed using the STRING and Cytospace software, and the top 9 hub genes identified were , , , , , , , , and .
Patients with LUAD can be clustered into three subtypes based on the expression of gene sets. These findings contribute to understanding the pathogenesis and molecular mechanisms in LUAD, and may lead to potential individualized pharmacogenetic therapy for patients with LUAD.
肺腺癌(LUAD)是肺癌的一种亚型,发病率和死亡率都很高。虽然基因分型是LUAD患者预后的一个重要决定因素,但关于基于基因集表达(GSE)分型的LUAD研究却很少。本研究采用GSE方法对LUAD患者进行基因分型。
从癌症基因组图谱(TCGA)和基因表达综合数据库(GEO)下载LUAD患者的临床和基因组信息。根据临床和基因集表达特征,将LUAD患者聚类为不同的分子亚型。比较各分子亚型之间的生存率和轮廓宽度。使用Kaplan-Meier生存曲线分析基因集之间生存率的差异。基于TCGA数据库,采用Cox回归和Lasso回归建立预后基因集模型,并使用GEO数据集进行验证。
最终共鉴定出10个核心基因,并聚类为3个亚型,平均轮廓宽度为0.96。这3个亚型的生存率存在显著差异(P<0.05)。基因本体(GO)分析表明,相关的生物学过程(BP)主要涉及细胞周期调控、有丝分裂细胞周期阶段转换和蛋白酶体介导的泛素依赖性蛋白分解代谢过程。细胞成分(CC)与纺锤体、染色体区域和中间体有关。分子功能(MF)主要集中在泛素样蛋白连接酶结合、翻译调节活性和氧化活性。京都基因与基因组百科全书(KEGG)分析表明,主要途径包括神经发生的爱泼斯坦-巴尔病毒感染途径、p53信号通路和蛋白质组途径。此外,使用STRING和Cytospace软件分析蛋白质-蛋白质相互作用网络,确定的前9个核心基因分别是 、 、 、 、 、 、 、 和 。
基于基因集的表达,LUAD患者可聚类为3个亚型。这些发现有助于理解LUAD的发病机制和分子机制,并可能为LUAD患者带来潜在的个体化药物遗传学治疗。