Wang Xiaotao, Luan Yu, Yue Feng
Department of Biochemistry and Molecular Genetics, Feinberg School of Medicine Northwestern University, Chicago, IL, USA.
Robert H. Lurie Comprehensive Cancer Center of Northwestern University, Chicago, IL, USA.
Sci Adv. 2022 Jun 17;8(24):eabn9215. doi: 10.1126/sciadv.abn9215. Epub 2022 Jun 15.
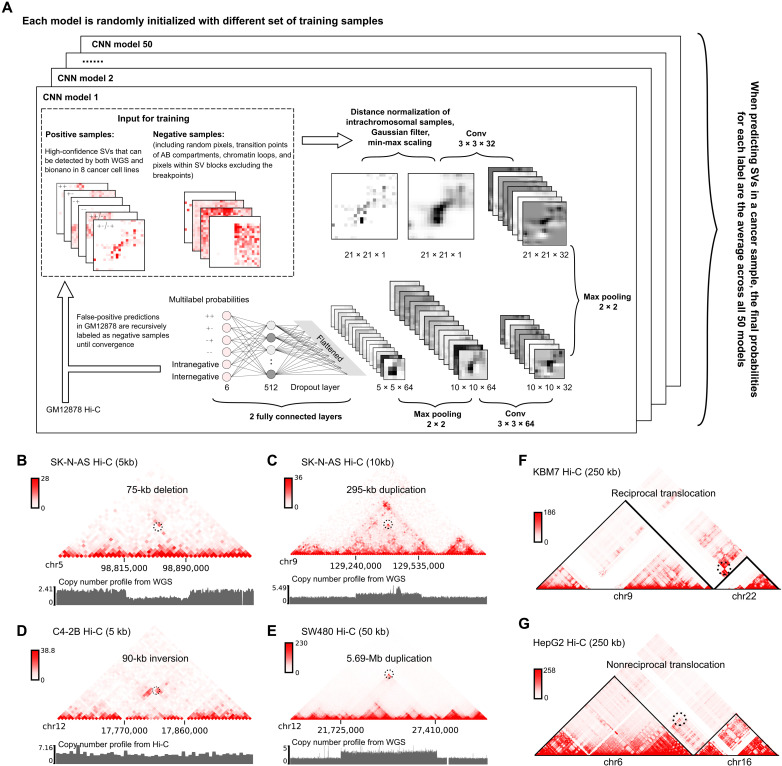
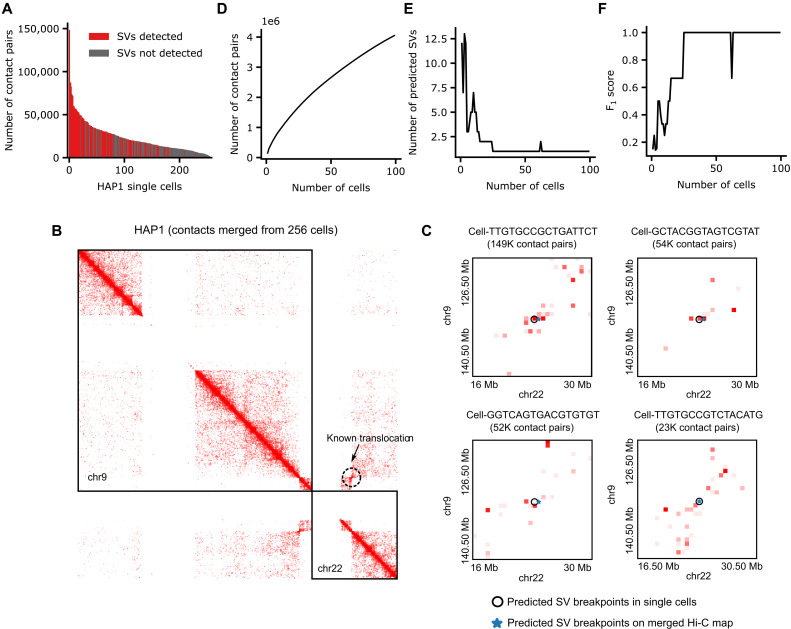
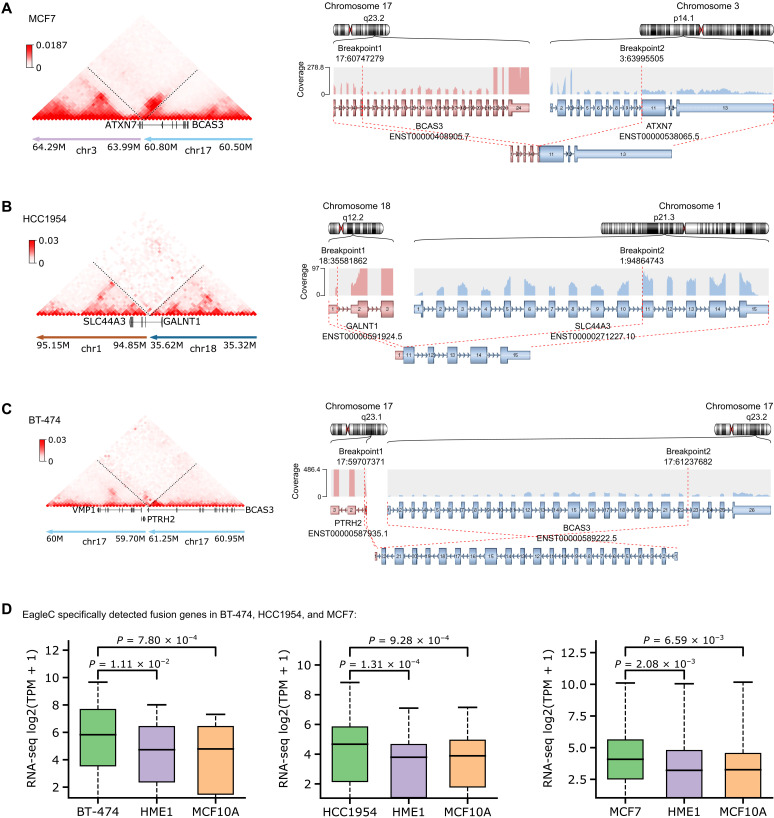
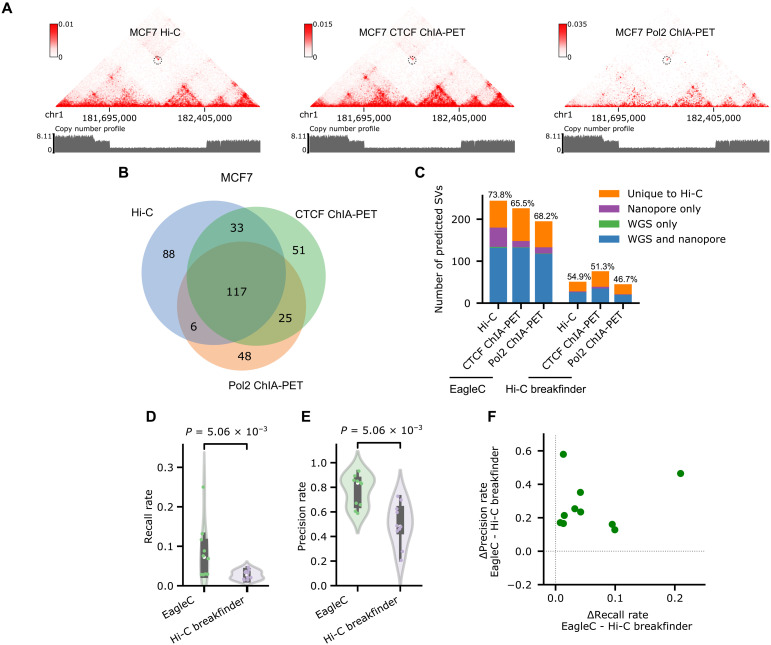
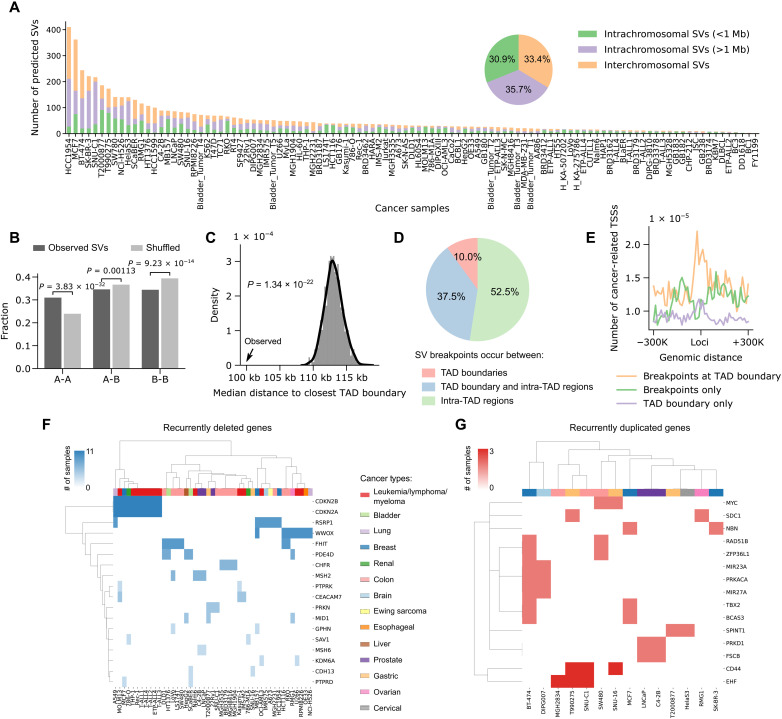
The Hi-C technique has been shown to be a promising method to detect structural variations (SVs) in human genomes. However, algorithms that can use Hi-C data for a full-range SV detection have been severely lacking. Current methods can only identify interchromosomal translocations and long-range intrachromosomal SVs (>1 Mb) at less-than-optimal resolution. Therefore, we develop EagleC, a framework that combines deep-learning and ensemble-learning strategies to predict a full range of SVs at high resolution. We show that EagleC can uniquely capture a set of fusion genes that are missed by whole-genome sequencing or nanopore. Furthermore, EagleC also effectively captures SVs in other chromatin interaction platforms, such as HiChIP, Chromatin interaction analysis with paired-end tag sequencing (ChIA-PET), and capture Hi-C. We apply EagleC in more than 100 cancer cell lines and primary tumors and identify a valuable set of high-quality SVs. Last, we demonstrate that EagleC can be applied to single-cell Hi-C and used to study the SV heterogeneity in primary tumors.
Hi-C技术已被证明是一种检测人类基因组结构变异(SVs)的有前景的方法。然而,严重缺乏能够利用Hi-C数据进行全范围SV检测的算法。当前方法只能以次优分辨率识别染色体间易位和长距离染色体内SVs(>1 Mb)。因此,我们开发了EagleC,这是一个结合深度学习和集成学习策略以高分辨率预测全范围SVs的框架。我们表明,EagleC能够独特地捕获一组全基因组测序或纳米孔测序遗漏的融合基因。此外,EagleC还能有效地捕获其他染色质相互作用平台中的SVs,如HiChIP、配对末端标签测序染色质相互作用分析(ChIA-PET)和捕获Hi-C。我们将EagleC应用于100多个癌细胞系和原发性肿瘤,识别出一组有价值的高质量SVs。最后,我们证明EagleC可应用于单细胞Hi-C,并用于研究原发性肿瘤中的SV异质性。